2015 Annual Science Report
 University of Illinois at Urbana-Champaign
Reporting | JAN 2015 – DEC 2015
University of Illinois at Urbana-Champaign
Reporting | JAN 2015 – DEC 2015
Project 7: Mining Archaeal Genomes for Signatures of Early Life: Comparison of Metabolic Genes in Methanogens
Project Summary
Methanogens represent the largest diversity among the archaea and have the unique ability to generate methane from simple compounds such as carbon dioxide, acetate and methylamines which were common in the anaerobic environments of early Earth and perhaps Mars. Methane biosynthesis also requires the presence/uptake of important ions such as sulfates, sulfides, carbonates, phosphates, and various light metal ions. In this project, we are attempting to analyze the evolution of the methanogens’ central cellular functions of translation, transcription, replication, and metabolism. To accomplish this, we are constructing the metabolic and regulatory networks of Methanosarcina acetivorans, the most complex methanogen known, and using these models to establish a framework for studying the evolution of methanogens. Results will be tested through microfluidic studies using varying carbon and ion sources.
Project Progress
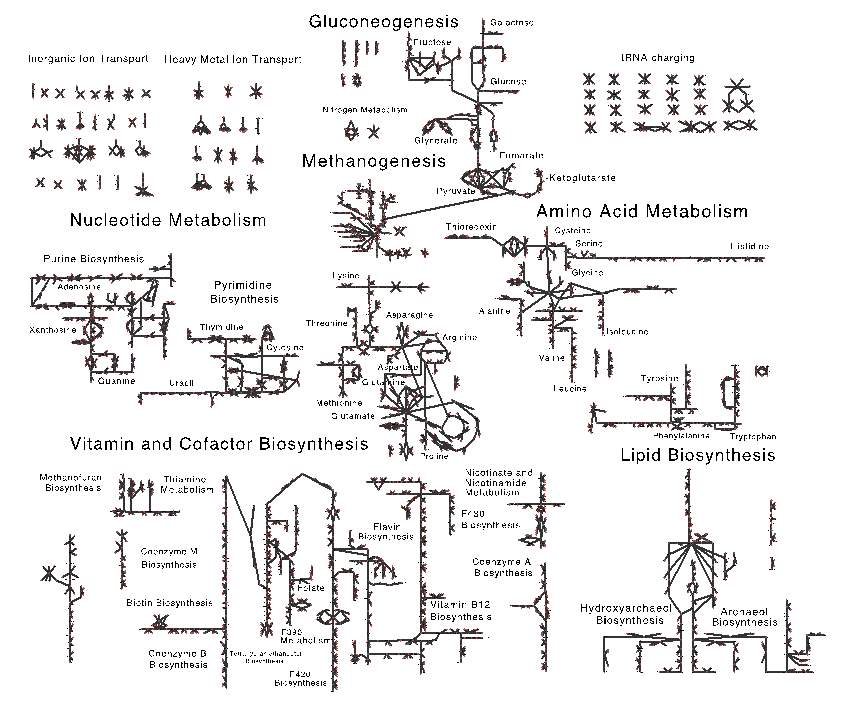
In our first methanogen paper [1] published in the journal Archaea, we focused primarily on the methanogenesis pathway. During the past year, we focused on developing a fully functional and complete metabolic and regulatory model for Methanosarcina acetivorans capable of capturing the metabolic fluxes observed under different growth enviornments. To do this, we build upon the work from Benedict, et al. [2] who had previously constructed a genome-scale metabolic model for M. acetivorans that contained 825 reactions associated with 745 genes. Our first step was to create a map of metabolism which allows, for the first time, visualization of metabolic fluxes, gene expression data, and evolutionary conservation of metabolic pathways in an integrative way. Our current map and the associated metabolic model (shown in Figure 1) serve as an invaluable tools to analyze metabolic behavior that results from the various enviornments in which the organism has evolved and will both be released with our currently submitted article.
During the map generation process we identified several key areas where the model could be improved. First, the pathway for pyrrolysine biosynthesis was added to reflect the requirement for this amino acid when growing on methylamine substrates. This amino acid is unique to methanogens, providing an interesting evolutionary case study. Second, we added the alternate cysteine aminoacylation pathway—a relic from more ancient methanogens—wherein O-phosphoserine is converted to cysteine while charged to tRNACys. Third, the methanofuran biosynthesis pathway, a unique cofactor found only in methanogens, was updated based on recent experimental characterization.
The model was refined by including organic osmolytes, salts and metals; factors that would have varied significantly over Earth’s history and have played key roles in defining how the methanogen adapted to its enviornments. By including osmolytes such as α-glutamate, N-acetyl-β-lysine and glycine betaine, which the cell produces in significant quantities to survive in their saline environments, we can probe the energetic requirements of maintaining osmotic pressure. By incorporating metals, we can investigate how changing metal availability, such as nickel, could have affected the prevalence of these oranism, and contributed to events such as the end-Permian extinction, for which methanogens have been implicated in causing. Altogether the new metabolic reconstruction consists of 757 reactions (827 including metabolite exchange) with 752 associated genes and greatly increases the predictive power of the model.
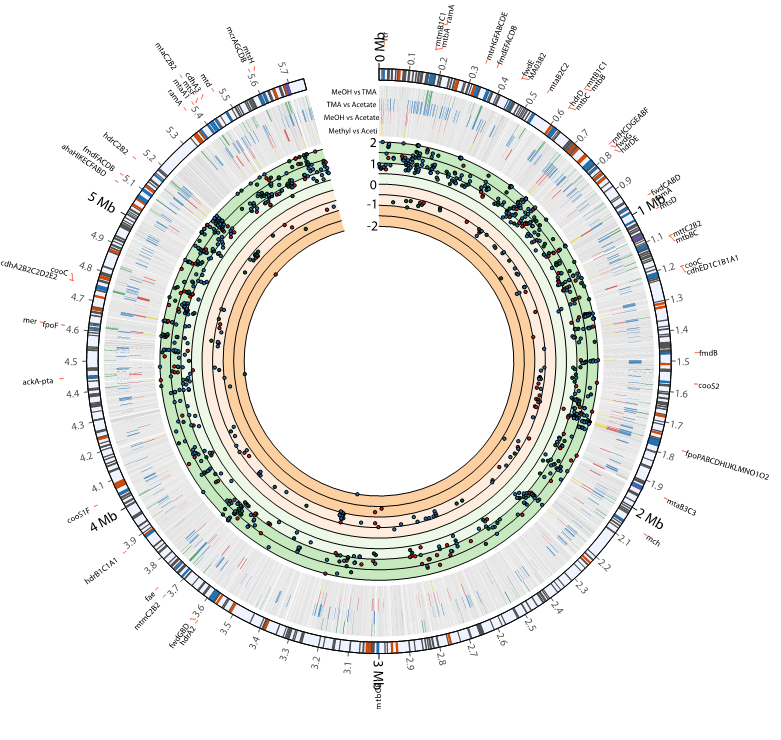
With this updated metabolic network, we used our mRNA half-life and expression data (Figure 3) to probe how this mRNA stability controls metabolic activity. This was done by classifying genes as either controlled by a transcriptional or degradational mechanism when comparing their expression across different growth substrates. The expression data was also integrated with the updated metabolic network for M. acetivorans to predict changes in pathway flux due to changes in carbon source, generating numerous hypothesis to be experimentally validated. We linked these metabolic changes to specific reactions and identified the mechanism of regulation predicted (transcription or degradation). The results suggest that M. acetivorans employs extensive post-transcriptional degradation mechanism along with less prevalent transcriptional regulation to optimize key metabolic steps for growth in different environments, and that degradation could play a much greater role in optimizing an organism’s metabolism than previously thought. These results have been submitted for review [3].
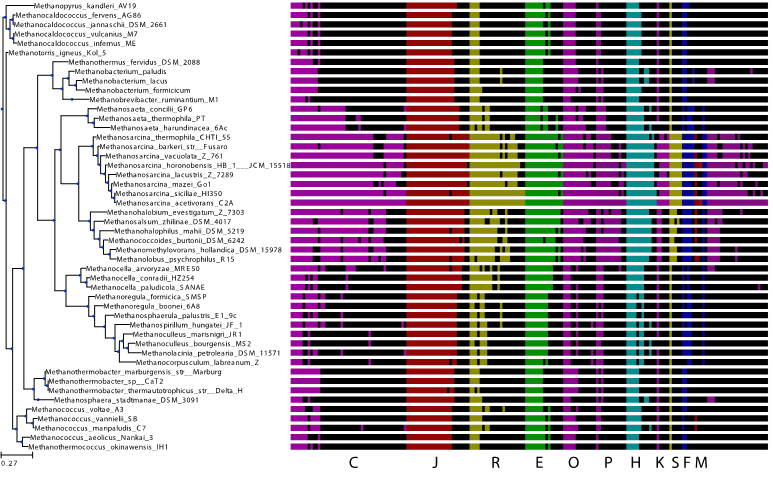
Using M. acetivorans as a model organism and our knowledge of metabolism, regulation, translation, our RNAseq data, and the ITEP database [4] of methanogen genomes, we identified the homologs of differentially expressed genes across the tree of methanogens as shown in Fig. 2. This genome-wide analysis covers all the cellular networks. The identification of these genes serves as the first analysis of regulation across methanogens and a unique perspective into the evolution of regulation in these ancient organisms. Ongoing work focuses on constructing integrated metabolic/regulatory networks for all methanogens which will allow us to identify how these organisms adapted to their varied environmental niches.
[1] Joseph R. Peterson, Piyush Labhsetwar, Jeremy R. Ellermeier, et al., “Towards a Computational Model of a Methane Producing Archaeum,” Archaea, vol. 2014, Article ID 898453, 18 pages, 2014. doi:10.1155/2014/898453.
[2] M. N. Benedict, M. C. Gonnerman, W. W. Metcalf, and N. D. Price. J. Bacteriol. 2012, 194(4):855.
[3] J.R. Peterson, S. Thor, L. Kohler, P.R.A. Kohler, W.W. Metcalf, Z. Luthey-Schulten, “mRNA Degradation Plays a Substantial Role in Regulating Metabolism in Methanosarcina acetivorans”, Submitted, 2016.
[4] M. N. Benedict, J. R. Henriksen, W. W. Metcalf, R. J. Whitaker, and N. D. Price, “ITEP: An integrated toolkit for exploration of microbial pan-genomes”, BMC Genomics, 2014, 15:8.
Publications
-
Peterson, J. R., Thor, S., Kohler, L., Kohler, P. R. A., Metcalf, W. W., & Luthey-Schulten, Z. (2016). Genome-wide gene expression and RNA half-life measurements allow predictions of regulation and metabolic behavior in Methanosarcina acetivorans. BMC Genomics, 17(1), None. doi:10.1186/s12864-016-3219-8
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Joseph Peterson
Co-Investigator
Shen Shee Thor
Co-Investigator
Isaac Cann
Collaborator
Bruce Fouke
Collaborator
Willam Metcalf
Collaborator
Rachel Whitaker
Collaborator
-
RELATED OBJECTIVES:
Objective 1.1
Formation and evolution of habitable planets.
Objective 2.1
Mars exploration.
Objective 3.1
Sources of prebiotic materials and catalysts
Objective 3.2
Origins and evolution of functional biomolecules
Objective 3.3
Origins of energy transduction
Objective 3.4
Origins of cellularity and protobiological systems
Objective 4.1
Earth's early biosphere.
Objective 4.2
Production of complex life.
Objective 5.1
Environment-dependent, molecular evolution in microorganisms
Objective 5.2
Co-evolution of microbial communities
Objective 5.3
Biochemical adaptation to extreme environments
Objective 6.1
Effects of environmental changes on microbial ecosystems
Objective 6.2
Adaptation and evolution of life beyond Earth
Objective 7.1
Biosignatures to be sought in Solar System materials
