2014 Annual Science Report
 University of Wisconsin
Reporting | SEP 2013 – DEC 2014
University of Wisconsin
Reporting | SEP 2013 – DEC 2014
Project 1E: Microbial Communities in Chocolate Pots Hot Spring, Yellowstone National Park
Project Summary
DNA was extracted from samples obtained from cores collected at six locations along a transect following the main fluid flow path at at Chocolate Pots (CP) hot spring, Yellowstone National Park. 454 pyrosequencing of 16S rRNA gene amplicons was performed on the extracts, resulting in the generation more than 70 amplicon libraries, each containing a between ca. 2500 and 7500 ca. 300 base pair-long reads. The raw reads were processed and analyzed for their phylogenetic affiliation and other comparisons using the QIIME pipeline. The results indicate that microbial communities in the upper few cm of the Fe/Si-rich CP deposits varied significantly along the sampling transect. Communities at two sites most proximal to the vent source differed substantially from one another and from communities at downstream sites. Although communities at downstream sites were not identical, they were more similar to one another than to the vent-proximal sites. A wide diversity of prokaryotic taxa, including both Bacteria and Archaea, were identified in the libraries, many of which are only distantly (e.g. <90% similarity in 16S rRNA gene sequence) related to known taxa. Communities in cores close to the vent were dominated by anaerobic taxa, many of which have the potential to function as Fe(III) reducers. This result is consistent with the relatively high abundance of reduced (ferrous) iron [Fe(II)] and the rapid rate of Fe(II) production observed in in vitro Fe(III) reduction experiments with material from sites near the vent. Abundant taxa at downstream sites included organisms related to the known Fe(II)-oxidizing organism Sideroxydans paludicola. These results are consistent with Fe geochemical data, which indicate that Fe(II) oxidation is likely the dominant Fe redox cycling pathway in deposits more than 1-2 meters from the vent source. A detailed metagenomic analysis of communities in the upper 1 cm at three sites is underway, with the goal of confirming the function of recognized taxa, and revealing the identity and function of potentially novel Fe redox cycling taxa.
Project Progress
Iron (Fe) biogeochemical cycling in circumneutral pH systems is an increasingly important astrobiological target in light of recent discoveries on Mars by Curiosity (Grotzinger et al., 2014). Previous research into microbial ferric iron [Fe(III)] reduction in Yellowstone National Park geothermal springs has focused on high temperature, low pH environments where microbial communities are able to utilize soluble Fe(III) as an electron acceptor for respiration (e.g. Kozubal et al., 2012). Much less attention has been paid to Fe(III)-reducing microbial communities in lower temperature, circumneutral pH environments, where solid phase Fe(III) oxides are the dominant forms of ferric iron.
This study represents the first broad-scale analysis of microbial communities in the warm (ca. 40-60 C), circumneutral pH (ca. 6.0-6.5) Chocolate Pots hot springs (CP) in Yellowstone National Park. The work was conducted in parallel with Fe biogeochemical analyses on the same set of cores described in the companion 2014 project report entitled “Iron biogeochemistry in Chocolate Pots hot spring, Yellowstone National Park” (referred to hereafter as the “companion report”). The central goal of the study was to obtain a phylogenetic characterization of in situ microbial communities through 454 pyrosequencing of 16S rRNA gene amplicons, and to relate community structure to in situ Fe biogeochemical conditions.
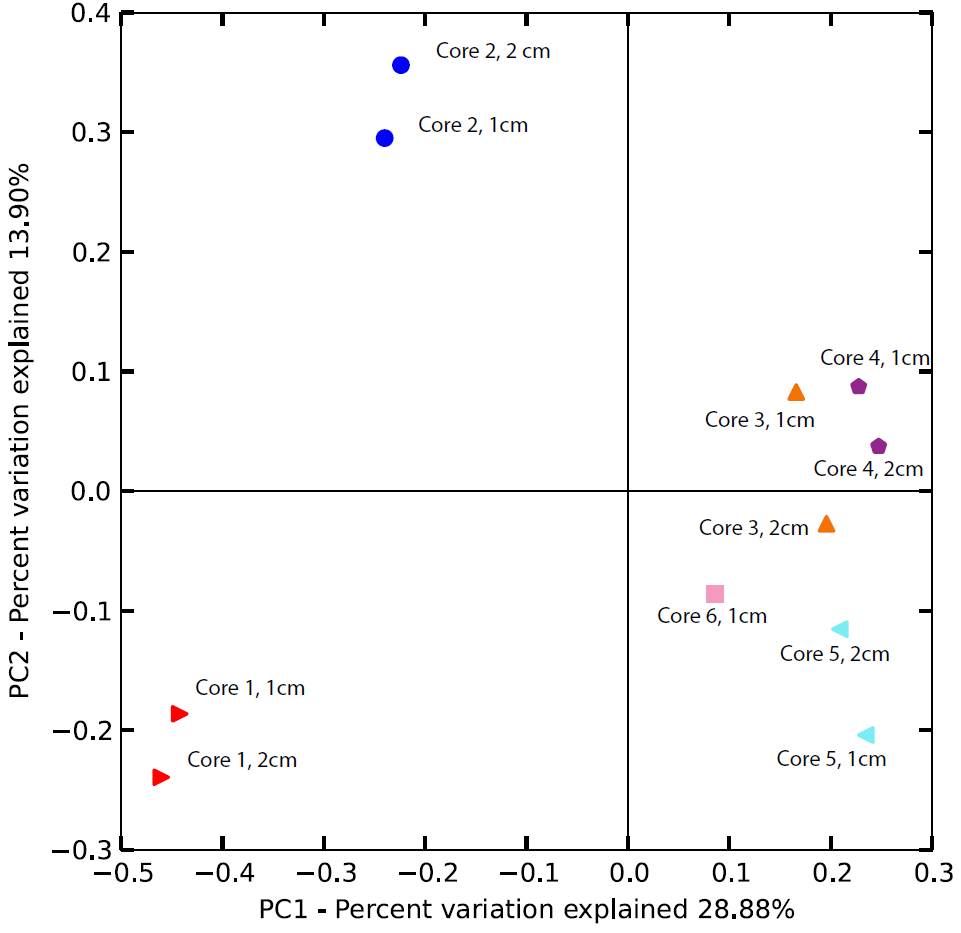
DNA was extracted from samples obtained from small cores collected at six locations along a transect following the main fluid flow path at CP, extending from the vent outlet to within a few meters of Gibbon River into which the hot spring discharges (see Fig. 1 in the companion report). The cores were sectioned at 1 cm intervals, and DNA was extracted using standard commercial (MoBio) kits. 454 pyrosequencing of 16S rRNA gene amplicons was conducted using previously described methods (Percak-Dennett and Roden, 2014). A total of 71 amplicon libraries were generated, each containing a total of between ca. 2500 and 7500 ca. 300 base pair-long “reads”. The raw reads were processed and analyzed for their phylogenetic affiliation using the QIIME pipeline (Caporaso et al., 2010), employing the Ribosomal Database Project (RDP) classifier algorithm (Wang et al., 2007). The principal component analysis (PCoA) facility within QIIME was used to compare selected libraries. For the sake of simplicity, we focus here on communities from the 0-1 cm and 1-2 cm sample depths, as well as communities from the in vitro Fe(III) reduction experiments described in the companion report, which were conducted with materials from the upper 2 cm of the deposit.
The results indicate that microbial communities in the upper few cm of the Fe/Si-rich CP deposits varied significantly along the sampling transect (Fig. 1). Communites at sites 1 and 2, i.e. those most proximal to the vent source, differed substantially from one another and from communities at downstream sites. Although communites at sites 3-6 were not identical, they were more similar to one another than to either site 1 or site 2.
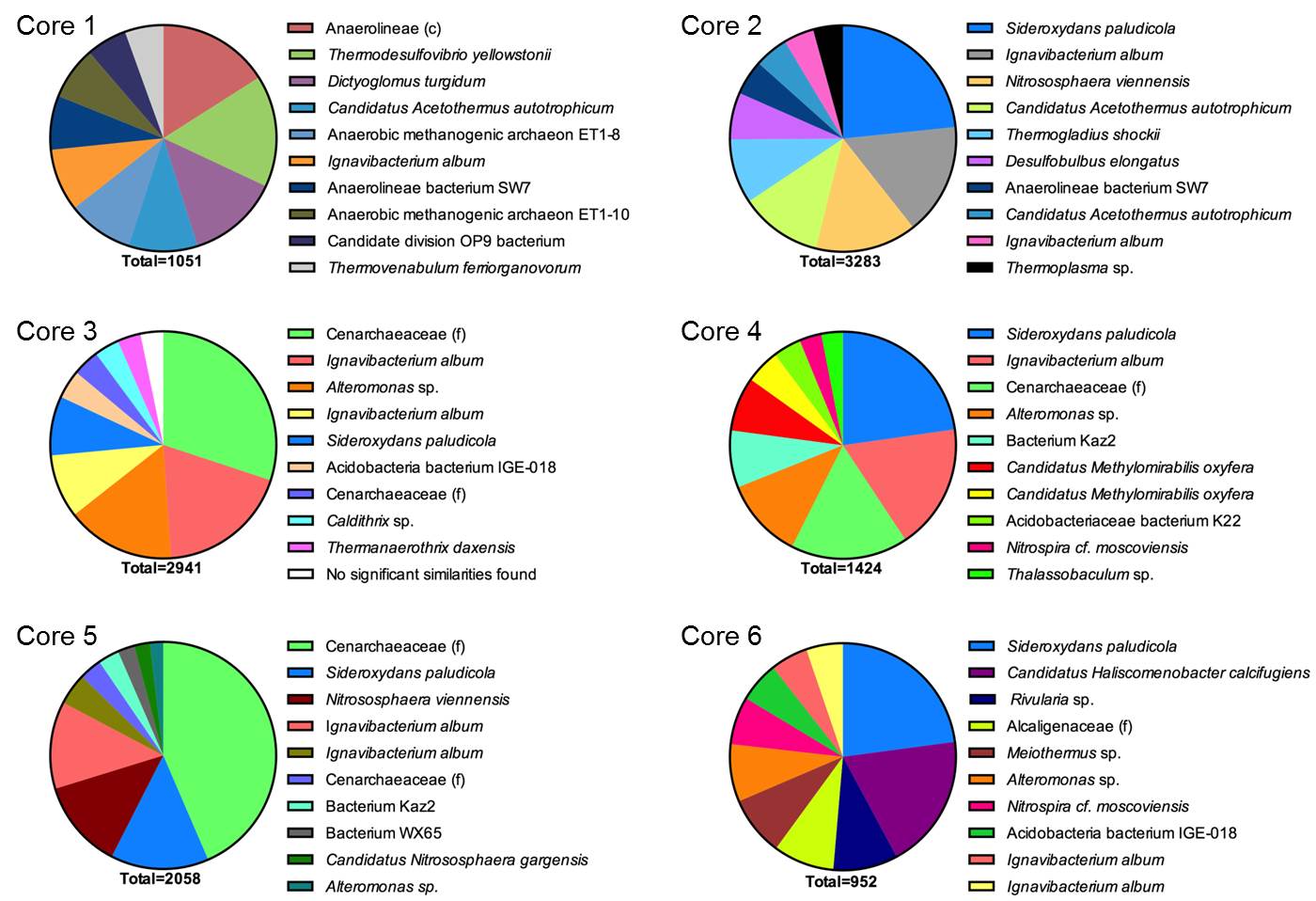
The RDP-generated phylogenetic assignments were supplemented with independent BLAST (Altschul et al., 1997) searches, which in most cases provided more specific information on a given read’s phylogenetic affiliation. Assignments for the top 10 most abundant reads for the 0-1 cm depth interval at each core sampling site are shown in Fig. 2. Although a detailed analysis of these data is beyond the scope of this brief report, a few key preliminary inferences can be made. First and foremost, it is important to note the wide diversity of prokaryotic taxa, including both Bacteria and Archaea, many of which are only distantly (e.g. <90% similarity in 16S rRNA gene sequence) related to known taxa. In due course the potential in situ function of these diverse, novel taxa will be assessed through metagenomic sequence analysis (see below). Second, the results clearly suggest that communities in core 1 were dominated by anaerobic taxa, many of which have the potential to function as Fe(III) reducers. For example, ca. 3 % of all reads (10-15% of the top 10 most abundant reads) were closely related to Ignavibacterium album, an organism which itself is closely related to a moderately thermophilic Fe(III) reducer recently isolated from anoxic subsurface fluids from an oil exploration well in Siberia (Podosokorskaya et al., 2013). This result is consistent with the relatively high abundance of reduced (ferrous) iron [Fe(II)] and the rapid rate of Fe(II) production observed in the in vitro Fe(III) reduction experiments with material from site 1 (see Figs. 2 and 3 in the companion report). Communities in core 2, which also contained significant quantities of Fe(II), were also dominated by anaerobic taxa. However, abundant core 2 taxa also included organisms related to the known Fe(II)-oxidizing organism Sideroxydans paludicola (Weiss et al., 2007). This same taxon was also abundant in cores 3-6 where, based on geochemical data, Fe(II) oxidation was likely the dominant Fe redox cycling pathway (see companion report).
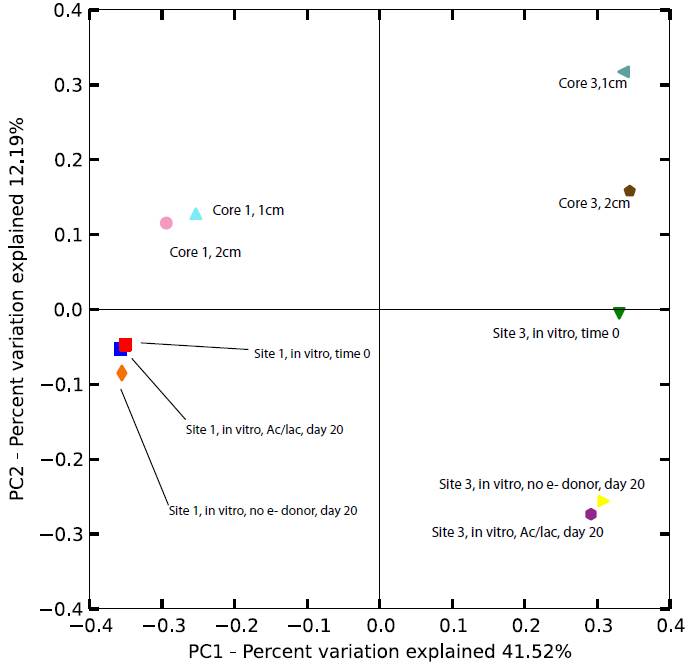
Microbial community composition in the upper 2 cm from cores 1 and 3 were compared to the initial (time zero) and 20-day samples from the in vitro Fe(III) reduction experiments conducted with materials from sites 1 and 3. The reason for doing this was to assess whether or not communities in materials where active Fe(III) reduction could be documented were analogous to those present in situ; if so, this would provide evidence (albeit indirect) that taxa detected in situ could be inferred to have Fe(III) reduction capacity. In general the results support this conclusion for the site 1: although PCoA revealed some differences in composition among the core vs. incubation experiment samples, variation was restricted compared to the large, first-order differences between taxa from sites 1 and 3 (Fig. 3). In addition, BLAST analyses indicated that many of the most abundant taxa in the core 1 samples were also abundant in the initial and 20-day samples from the incubation experiment (data not shown). The apparent significant shift in the 20-day incubation experiment samples for site 3, compared to the initial and core samples, may be attributed to the effect of imposing anaerobic, Fe(III)-reducing conditions on materials that were not experiencing those conditions in situ. Consistent with this suggestion, BLAST analysis revealed that the relative abundance of Sideroxydans-related (i.e. Fe(II)-oxidizing) taxa decreased during anaerobic incubation, whereas the relative abundance of Ignavibacterium-related (i.e. Fe(III)-reducing) taxa increased.
In summary, this study provides the first detailed phylogenetic assessment of in situ microbial communities within the CP hot spring. Though still preliminary, our analysis of the 16S rRNA gene amplicon data suggests the presence of a wealth of novel bacterial and archaeal taxa. Although the function of many of these novel taxa remains to be determined, the presence of organisms related to known Fe(III)-reducing and Fe(II)-oxidizing taxa is consistent with an active Fe redox cycling system, as suggested by parallel geochemical data. We have acquired Illumina HiSeq metagenomic libraries for the 0-1 cm depth interval samples from cores 1, 2 and 3, with the goal of confirming the function of recognized taxa, and revealing the identity and function of potentially novel Fe redox cycling taxa. The assembled libraries have been submitted to JGI’s IMG/M on-line system (Markowitz et al., 2007) for phylogenetic and functional analysis, and we are in the process of binning the libraries using the newly-developed CONCOCT software (Alneberg et al., 2014). Detailed genomic analysis of reconstructed genomes (e.g. Wrighton et al., 2012; Wrighton et al., 2014) should ultimately provide a wealth of insight into the biochemical underpinnings of in situ Fe redox cycling in this unique geothermal environment.
References
Alneberg, J., B. S. Bjarnason, I. de Bruijn, M. Schirmer, J. Quick, U. Z. Ijaz et al. 2014. Binning metagenomic contigs by coverage and composition. Nat. Methods 11:1144-1146.
Altschul, S. F., T. L. Madden, A. A. Schaffer, J. H. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402.
Caporaso, J. G., J. Kuczynski, J. Stombaugh, K. Bittinger, F. D. Bushman, E. K. Costello et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335-336.
Grotzinger, J. P., D. Y. Sumner, L. C. Kah, K. Stack, S. Gupta, L. Edgar et al. 2014. A habitable fluvio-lacustrine environment at Yellowknife Bay, Gale Crater, Mars. Science 343:DOI: 10.1126/science.1242777.
Kozubal, M. A., W. P. Inskeep, and R. E. Macur. 2012. Geomicrobiology of iron oxide mats from acidic geothermal springs in Yellowstone National Park: Microbial community structure, isolation of novel species and mechanisms of iron oxidation. Front. Microbiol. 3:109.
Markowitz, V. M., E. Szeto, K. Palaniappan, Y. Grechkin, K. Chu, I. M. A. Chen et al. 2007. The integrated microbial genomes (IMG) system in 2007: data content and analysis tool extensions. Nucl. Acids Res.gkm846.
Percak-Dennett, E. M., and E. E. Roden. 2014. Geochemical and microbiological responses to oxidant introduction in reduced subsurface sediment from the Hanford 300 Area, WA. Environ. Sci. Technol. 48:9197-9204.
Podosokorskaya, O. A., V. V. Kadnikov, S. N. Gavrilov, A. V. Mardanov, A. Y. Merkel, O. V. Karnachuk et al. 2013. Characterization of Melioribacter roseus gen. nov., sp nov., a novel facultatively anaerobic thermophilic cellulolytic bacterium from the class Ignavibacteria, and a proposal of a novel bacterial phylum Ignavibacteriae. Environ. Microbiol. 15:1759-1771.
Wang, Q., G. M. Garrity, J. M. Tiedje, and J. R. Cole. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261-5267.
Weiss, J. V., J. A. Rentz, T. Plaia, S. C. Neubauer, M. Merrill-Floyd, T. Lilburn et al. 2007. Characterization of neutrophilic Fe(II)-oxidizing bacteria isolated from the rhizosphere of wetland plants and description of Ferritrophicum radicicola gen. nov. sp. nov., and Sideroxydans paludicola sp. nov. Geomicrobiol. J. 24:559-570.
Wrighton, K. C., B. C. Thomas, I. Sharon, C. S. Miller, C. J. Castelle, N. C. VerBerkmoes et al. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337:1661-1665.
Wrighton, K. C., C. J. Castelle, M. J. Wilkins, L. A. Hug, I. Sharon, B. C. Thomas et al. 2014. Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. Isme J. 8:1452-1463.
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Eric Roden
Project Investigator
Eric Boyd
Co-Investigator
Nathan Fortney
Co-Investigator
Brandon Converse
Collaborator
-
RELATED OBJECTIVES:
Objective 2.1
Mars exploration.
Objective 4.1
Earth's early biosphere.
Objective 5.1
Environment-dependent, molecular evolution in microorganisms
Objective 5.3
Biochemical adaptation to extreme environments
Objective 7.1
Biosignatures to be sought in Solar System materials