2014 Annual Science Report
 University of Illinois at Urbana-Champaign
Reporting | SEP 2013 – DEC 2014
University of Illinois at Urbana-Champaign
Reporting | SEP 2013 – DEC 2014
Project 7 Control of Evolvability and Chromosomal Rearrangement by Stress
Project Summary
Evolution of the genome happens predominantly by gross chromosomal rearrangements (GCR), usually involving non-homologous recombination, and point mutation or single nucleotide variation (SNV). GCR re-assorts domains and regulatory functions, and frequently changes gene copy number, allowing further evolution. SNV changes coding sequences, modifying the properties of encoded macromolecules and their regulation. We are able to measure both SNV and GCR in the same assay in Escherichia coli, and have established that both are dramatically up-regulated in response to stress. Through many year’s work, we have learnt many details of these mechanisms, but some of the outstanding questions are crucial to understanding the significance of stress in evolution, notably the signaling and execution of the pathway from stress to mutation, and how the decision is made between SNV and GCR. This project aims to answer these questions.
Project Progress
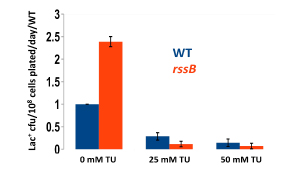
We have screened for suppressors of the requirement for the RpoS stress-response in chromosomal rearrangement by using a transposon with outward-facing promoters that activates genes in the absence of stress. This could tell which stress-induced function is required for chromosomal rearrangement. Most of the suppressors isolated have activated parts of cryptic prophages: bacterial viruses that have lost their ability to lyse the host cell, and so the genetic material lives on in an inactive form in the host cell’s DNA. Four of these suppressors have resulted in the activation of alternative recombination through the RecET pathway. We are testing whether these have bypassed the requirement for stress, rather than suppressing it. The most interesting isolate at present has the transposon disrupting the gene dgt, which encodes a deoxyguanidine triphosphohydrolase. This protein regulates both deoxyguanidine triphosphate and thymidine triphosphate pools, and is stress-induced. Other work has shown that DNA nucleotide pool content modulates mutation rates, but the mechanism for this is unknown. Work on validating and explaining the effect of dgt is ongoing.
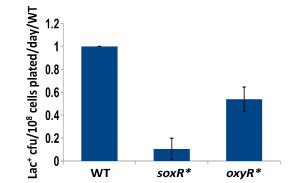
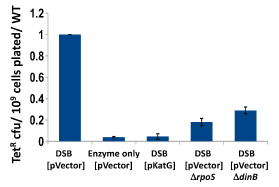
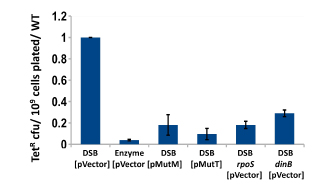
Nucleoid-associated proteins (NAPs) both modify the structure of bacterial chromatin and act as global regulators of gene expression. We have completed a study of the involvement of nucleoid-associated proteins in stress-induced mutation, finding that the effects of four NAPs are explained by factors other than their ability to modify chromatin structure. Two of the four modify the availability of oxygen radicals (see below), another regulates the expression of the DNA damage response, specifically the availability of trans-lesion polymerases, and the fourth is required for maintenance of the conjugative plasmid that carries the assay system. Thus we find no evidence from these experiments that chromatin remodeling regulates mutagenicity under stress.
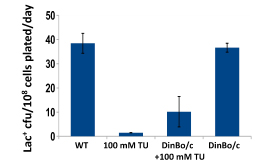
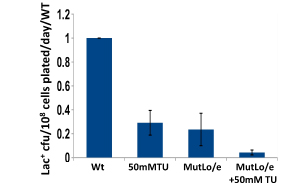
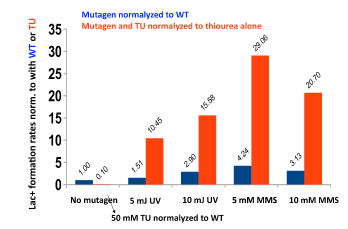
We have established the unexpected result that both spontaneous mutation and chromosomal rearrangement require 8-oxoguanine incorporation into DNA. We already knew that double-strand breaks are required, but the requirement for oxidative DNA lesions is not via double-stand break formation. Using genetic control of responses to DNA damage via oxidation and using chemical scavengers of reactive oxygen, we show beyond doubt that there is a requirement for persistence of incorporated 8-oxoguanine. When we over-express enzymes that specifically excise 8-oxoguanine from DNA, point mutation formation is deficient. Hence the lesion must remain in the DNA to be effective, and so mutation is presumably formed by induction of trans-lesion synthesis. We have not yet shown that chromosomal rearrangement requires the lesion to remain in DNA, but are setting up a new assay to test that. Because reactive oxygen will not be present in all environments, we tested whether other DNA lesions could substitute for 8-oxoguanine. Suppressing reactive oxygen with thiourea, a condition that allows no mutation, we have shown that we can restore mutation with alkylating agents or with ultraviolet irradiation. Genetic tests indicate that these treatments restored the same stress-dependent pathway that we study. We have formulated a model in which base lesions in DNA are required to pause the highly processive replicative polymerase, allowing alternative error-prone polymerases to attain the active site on the replisome.
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Philip Hastings
Project Investigator
Susan Rosenberg
Co-Investigator
Jessica Moore
Collaborator
Philip Thornton
Collaborator
-
RELATED OBJECTIVES:
Objective 5.1
Environment-dependent, molecular evolution in microorganisms