2013 Annual Science Report
 University of Illinois at Urbana-Champaign
Reporting | SEP 2012 – AUG 2013
University of Illinois at Urbana-Champaign
Reporting | SEP 2012 – AUG 2013
Control of Evolvability and Chromosomal Rearrangement by Stress
Project Summary
Gross chromosomal rearrangements (GCRs) underlie much of evolution, changing the copy number of genes, allowing development of new functions by providing redundant genetic material, reassorting protein domains and reassorting regulatory elements. Some mechanisms of chromosomal rearrangement are understood, but most are not. Using a model system in Escherichia coli, we have shown that both point mutation and GCRs occur preferentially when the cells are stressed, and require several stress-responses to be activated. We seek to understand the regulation of GCR by discovering how stress regulates the process, and what is the decision that activates the GCR pathway rather than a parallel stress-induced point mutation pathway. These are important components of the mechanisms by which organisms evolve to adapt to new or changing environments.
Project Progress
Using the lac assay in Escherichia coli we are screening for suppression of the requirement for the general and starvation stress-response regulator, RpoS, for gross chromosomal rearrangement (GCR). Using transposon mutagenesis with outward-facing promoters allows us to screen for loss of function by disruption of a gene, or for up-regulation by insertion upstream of a gene. The lac assay measures the frequency of cells that mutate to lactose utilization either by point mutation or by amplification of the leaky allele. Amplification is a proxy for GCR. The initial screen of about 15,000 transposon insertions yielded 200 apparent suppressors of stress-induced mutation requirement for RpoS. These were subjected to a secondary screen using re-version of a tetracycline sensitivity gene that cannot be reverted by GCR. There were 89 clones that did not revert in the tet assay. These strains have potentially activated an RpoS regulated gene that is required for GCR, so that identifying them might tell us how RpoS regulates GCR, and hence how stress regulates genomic change. We have now screened all 89 these, and confirmed that ten of them do not require RpoS for amplification. The genes disrupted or activated by the transposon were sequenced. Seven suppressed clones carry transposon insertions that activate functions of cryptic prophage. These do not require Pol I or RpoS for amplification, and therefore represent activation of an alternative pathway that is not stress-induced. One insertion disrupts pabA, involved in nucleotide synthesis, while another activates dgt, a known mutator gene concerned in regulation of nucleotide pools. We do not yet know how these gene products regulate genomic change. One insertion is not yet resolved. Discounting the prophage insertions, we see that the screen is not saturated. We are therefore setting up to run the mutant screen again to get closer to saturation and find further genes.
Using the same lac assay, we are probing the involvement of nucleoid associated proteins (NAPs) in stress-induced mutation. NAPs condense DNA, limiting access of DNA-acting proteins, namely proteins concerned in regulation, transcription, replication and repair. They are important in controlling the response of the genome to stressful environments. We find that 11 of 14 NAPs are required for both GCR and point mutation, showing that they do not control choice of mutation pathway. One NAP, Dps, inhibits mutation, while two are not involved.
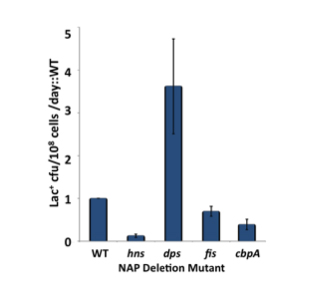
For 4 of the 12, we have confirmed that they are involved in the same pathways of genetic change that we have been studying, by their epistatic interactions with rpoS and recA.
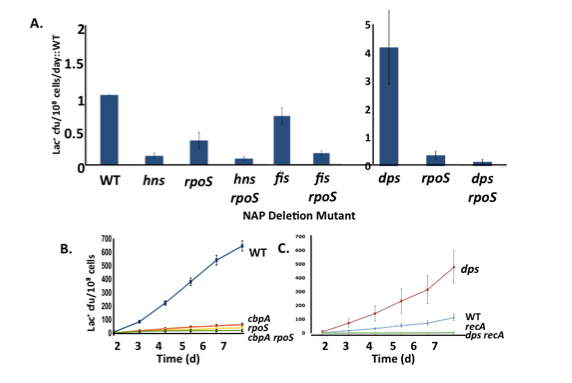
We are probing in detail the actions of dps, fis, hns and cbpA. We have shown that the role of Dps is to scavenge reactive oxygen species (ROS) within the cell, thus revealing a previously unsuspected involvement of ROS in stress-induced mutation.
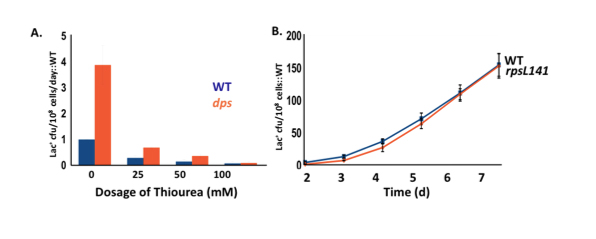
Dps sequesters iron, thus limiting the Fenton reaction that generates ROS, and it also scavenges oxygen radicals and sequesters them away from DNA and proteins. Dps is strongly expressed in starving cells, co-crystallizing with DNA. We also find that protein oxidation damage is not involved (Fig. 3). Fis and H-NS are mostly expressed during growth. We see that strains deleted for fis and H-NS have defects in homologous recombination (required for stress-induced mutation), but are not sensitive to ultraviolet light (CbpA has no defect in either). fis and H-NS deletions also have defects in DNA damage (SOS) response induction.
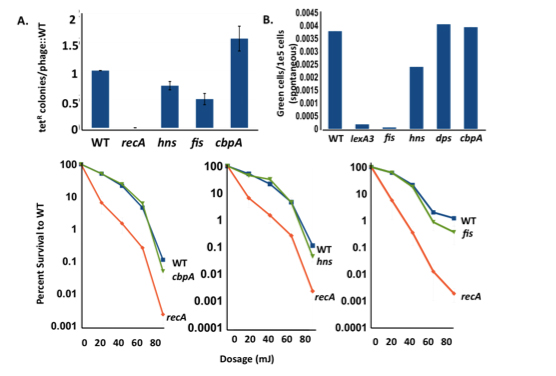
They are epistatic with RpoS, showing that they work in the same pathway as the stress response regulator. We are investigating the possibility that they work through regulation of genes upstream of RpoS in the sensing and transduction of signals of starvation. A CbpA mutant, an orthologue of Dps, is strongly down for amplification and point mutation in contrast to Dps mutants. This work continues.
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Philip Hastings
Project Investigator
Susan Rosenberg
Co-Investigator
Jessica Moore
Collaborator
Philip Thornton
Collaborator
-
RELATED OBJECTIVES:
Objective 5.1
Environment-dependent, molecular evolution in microorganisms