2012 Annual Science Report
 Georgia Institute of Technology
Reporting | SEP 2011 – AUG 2012
Georgia Institute of Technology
Reporting | SEP 2011 – AUG 2012
Resurrection of an Ancestral Peptidyl Transferase
Project Summary
Ancient components of the ribosome, inferred from a consensus of previous work, were constructed in silico, in vitro, and in vivo. The resulting model of the ancestral ribosome presented here incorporates about 20% of the extant 23S rRNA and fragments of four ribosomal proteins. We test hypotheses that ancestral rRNA can: (i) assume canonical 23S rRNA-like secondary structure, (ii) assume canonical tertiary structure, and (iii) form native complexes with ribosomal protein fragments. Footprinting experiments support formation of predicted secondary and tertiary structure. Gel shift, spectroscopic and yeast three-hybrid assays show specific interactions between ancestral rRNA and ribosomal protein fragments, independent of other, more recent, components of the ribosome. This robustness suggests that the catalytic core of the ribosome is an ancient construct that has survived billions of years of evolution without major changes in structure. Collectively, the data here support a model in which ancestors of the large and small subunits originated and evolved independently of each other, with autonomous functionalities.
Project Progress
The ribosome is a molecular machine that synthesizes all coded proteins. It is made of a small subunit (SSU) that decodes messenger RNA and a large subunit (LSU) that catalyzes formation of peptide bonds. Some components of the ribosome are highly conserved throughout extant life (1) and are considered to be among the oldest structures in biology (2-7). The peptidyl transferase center (PTC), for example, is thought to predate coded protein (8-12). If so, the PTC emerged from an ancient biological world, possibly an ‘RNA World’ (13-17), before life adopted all the processes of Crick’s “central dogma” (18). In this scenario the PTC was an active participant in the origins of current biology and is one of our most direct biochemical links to the distant evolutionary past.
Several groups, including Fox (11), Noller (12), Steinberg (8), Williams (9), and Gutell and Harvey (1), have proposed molecular-level events in early ribosomal evolution or have determined universally conserved ribosomal components (Figure 1). These proposed evolutionary pathways can be used to predict specific sequences and structures of ancestral rRNAs and polypeptides. Here we use a consensus of proposed evolutionary pathways and of conserved ribosomal components (Figure 1) to design and construct a molecular-level model, in silico, in vitro and in vivo, of an ancestral PTC (a-PTC, Figure 2). This model is intended as a starting platform for an iterative hypothesis-testing approach for understanding the origins of translation. Our design process relies substantially on three-dimensional structures, which are more conserved than sequence over long evolutionary time frames (9,19,20). The a-PTC incorporates fragments of the 23S rRNA, fragments of ribosomal proteins and divalent cations.
The ancestral rRNA fragments inferred here from consensus (Figure 1) are joined together to form a single RNA polymer (a-rRNA, Figure 2). The a-rRNA contains rRNA that forms and surrounds the PTC, which is composed of fragments from Domains II, IV and V. Using the 3D structure of the T. thermophilus LSU (21), the 23S rRNA was ‘shaved’ to a rough sphere of around 30 Å in radius, centered at the site of peptidyl transfer. This shaving process created 13 rRNA fragments. To facilitate re-connection of these fragments to form a polymer, termini were selected preferentially in A-form helical regions of the rRNA 3D structure and were capped with stem-loops. The result is a single RNA polymer containing the most ancient 20% of the T. thermophilus 23S rRNA. The fragments are stitched together by stem-loops in such a way that the 3D structure of the PTC will be maintained (Figure 2). The a-rRNA contains rRNA that is (i) universally conserved in secondary and three-dimensional structure in extant organisms and organelles (1,9), (ii) tightly networked by molecular interactions (11), (iii) densely coordinated by Mg2+ ions (22), and (iv) found in the central shells of the ribosomal ‘onion’. The ribosomal onion is a conceptual model in which the age of elements of the ribosome are considered to correlate inversely with radial distance from the site of peptidyl transfer (9).
Ribosomal proteins do not engage directly in catalytic processes in the ribosome (23,24). However, the non-globular ‘tails’ of some ribosomal proteins do penetrate the LSU and interact extensively with the rRNA that forms the catalytic PTC. The tails of ribosomal proteins L2, L3, L4, L15, and L22 penetrate the 30 Å sphere that defines the a-rRNA and are highly conserved in conformation throughout the three domains of life (25-27). We shaved those proteins at the 30 Å boundary used for the 23S rRNA, and included the resulting peptides in the a-PTC. The shaving process did not disrupt protein secondary structure; none of these protein tails are globular. The ancestral peptide components of the a-PTC are called a-rPeptide L2, a-rPeptide L3, a-rPeptide L4, a-rPeptide L15, and a-rPeptide L22 (see Table 1 for peptide sequences). It was previously proposed that these tails are more ancient than any protein with globular structure (9).
Using computation, in vitro and yeast-three-hybrid experiments, we test the hypotheses that: (i) a-rRNA adopts LSU-like secondary structure, as shown in Figure 2A, (ii) a-rRNA in association with Mg2+ adopts LSU-like tertiary interactions, as shown in Figures 2B and 3, and (iii) a-rRNA forms specific LSU-like complexes with a-rPeptides, as shown in Figure 2B. Components of the a-PTC were assembled in silico, in vitro, and in vivo and these hypotheses tested by various modeling, footprinting, and binding assays. A longer-range goal is determining if a-rRNA in association with a-rPeptides can form a functional a-PTC, as defined by catalytic activity.
a-PTC in silico
The a-PTC was modeled in three dimensions (Figure 2B). To form the in silico a-PTC model, the X-ray structure of the LSU of T. thermophilus (PDB 2J01) was shaved to a sphere of around 30 Å, and 12 of the 13 resulting rRNA fragments were stitched together with stem-loops, each of sequence 5’-gccGUAAggc-3’. An exception is the rRNA fragments adjacent to helices 47 and 61, where the rRNA fragments were joined directly, without a stem-loop (Figure 2A). The completed a-rRNA is composed of 505 nucleotides derived from the 23S rRNA plus 110 nucleotides from 11 stem-loops. The a-PTC contains a-rRNA (615 nucleotides), Mg2+ ions, and 5 a-rPeptides (Table 1).
In the 3D model, the components that are common between the a-PTC and the T. thermophilus LSU crystal structure have conserved conformation and interactions. The stem-loops are constrained to canonical GNRA tetraloop conformation (28) and are located on the surface of the a-PTC. The stem-loops do not engage in unfavorable steric contacts with other parts of the a-PTC. The model is stereochemically reasonable.
The predicted secondary structure of the a-PTC shown in Figure 2A is based on the canonical secondary structure of the 23S rRNA. There are some small differences between the canonical secondary structure and the actual secondary structure found in the crystal structure. These difference are responsible for some otherwise unexpected experimental observations, as discussed below.
RNase H characterization of a-rRNA secondary structure
RNase H and fifteen different DNA oligonucleotide probes were employed to characterize the secondary structure of a-rRNA in vitro (Figure 3A). RNase H cleavage is broadly used to characterize RNA secondary structure, allowing discrimination between single-stranded and double-stranded RNA (29,30). RNase H recognizes and cleaves RNA/DNA heteroduplexes; double-stranded RNA inhibits cleavage by blocking heteroduplex formation. The RNase H cleavage pattern of the a-rRNA shows good overall agreement with predictions of the in silico model, suggesting formation of the secondary structure shown in Figures 2A and 3. Four DNA probes were designed to target regions of a-rRNA that are double-stranded in the predicted secondary structure, and so should not yield RNase H cleavage. Eleven DNA probes were designed to target regions of a-rRNA that are single-stranded in the predicted secondary structure, which should yield RNase H cleavage. Ten of 11 predicted single-stranded RNA target regions are cleaved partially or fully by RNase H. Three of the 4 predicted double-stranded target regions are protected from cleavage, or showed only partial cleavage.
RNase H cleavage in the presence of two of the DNA probes initially appeared to suggest inconsistencies between the predicted and observed a-rRNA secondary structure, but further analysis indicated otherwise. Specifically, probe S36 targets RNA that is single-stranded in the predicted secondary structure, and therefore was anticipated to yield cleavage by RNase H. However, inspection of the T. thermophilus 50S crystal structure (21) reveals that the target region of probe S36 is engaged in secondary interactions not reflected in the canonical 23S secondary structure on which the predicted a-rRNA secondary structure is based. S36 does not yield cleavage of a-rRNA consistent with the secondary interactions observed in the LSU crystal structure.
Oligonucleotide D74 targets Helix 74, which is double-stranded in the predicted secondary structure, and thus D74 was not expected to yield cleavage. Contrary to this prediction, D74 yields significant cleavage (~88%) compared to the other double-stranded target regions. Comparing RNA/RNA vs DNA/RNA duplex free energies (31,32), we find that the stability of Helix 74 (-5.80 kcal/mol) is less than that of the DNA/RNA heteroduplex formed by D74 with the target RNA (-12.5 kcal/mol). This differential stability may allow the probe to invade the RNA/RNA duplex, accounting for the cleavage.
SHAPE characterization of a-rRNA secondary structure
SHAPE utilizes an electrophile, in this case NMIA, that reacts with 2’-hydroxyl groups of RNA (33). SHAPE reactivity is modulated by RNA flexibility. Base-paired nucleotides exhibit low reactivity since their flexibility is constrained. Single-stranded nucleotides are flexible and reactive to the electrophile. SHAPE has been shown to be accurate for mapping secondary structure of many RNAs, including E. coli rRNA (34).
The SHAPE data show excellent correspondence with the predicted secondary structure of a-rRNA (Figure 3A). These data were obtained by probing the a-rRNA in the presence of monovalent cations (250 mM Na+) and the absence of Mg2+. These conditions are known to support the formation of RNA secondary structure, but not tertiary structure (35-37). SHAPE data were obtained for 459 of the 505 nucleotides (91%) of the a-rRNA derived from the 23S (stem-loop nucleotides are analyzed separately below). The values and statistics provided in the remainder of this section refer only to these nucleotides derived from the 23S rRNA. Data were not obtainable for RNA near the termini, or for a few nucleotides with high background (i.e., high reverse transcriptase termination in samples not treated with NMIA). These regions are shown in gray on the a-rRNA secondary structure in Figure 3A.
Nucleotides were binned into four groups according to their normalized SHAPE reactivity. U2574 (NCBI E. coli numbering) is the most reactive nucleotide, with an absolute reactivity of 3.24 (arbitrary units), and is considered 100% reactive. Bin assignments are indicated by the size of red triangles in Figure 3A. The largest triangles denote the 13 nucleotides with normalized SHAPE reactivities ranging from 38-100%. Triangles of intermediate size indicate the 24 nucleotides with reactivities from 25-37%. The smallest triangles indicate the 109 nucleotides with reactivities from 8-24%. Nucleotides with reactivities of ≤ 7% are considered to be unreactive.
Of the 146 nucleotides that are reactive to the SHAPE reagent, 133 (91%) fall in regions expected to be single-stranded, in that they do not form base pairs in the predicted secondary structure. These nucleotides are in loops, bulges, or other single-stranded regions. The resolution of SHAPE is sufficiently high to allow identification of small bulges such as those in Helices 35, 61, 64, 73, 74, and 89 (Figure 3A). Thirteen reactive nucleotides are base-paired in the predicted secondary structure. These nucleotides represent less than 6% of the 232 nucleotides that are expected to be double-stranded, the remaining 94% of which exhibit low SHAPE reactivity as predicted. The 133 nucleotides that SHAPE identifies as flexible compose 59% of the 227 single-stranded nucleotides in the predicted secondary structure.
gccGUAAggc stem-loops fold in all contexts. SHAPE data were obtained for 106 (96%) of the 110 nucleotides within the 11 stem-loops. The stem regions are unreactive. Only 5 of the observable 62 nucleotides in the stem regions (8%) exhibit reactivity, which is low for each. The loop regions yield a consistent pattern of reactivity that is independent of the surrounding sequence. In 10 of the 11 stem-loops, the UAA nucleotides are more reactive than the preceding G. This pattern of reactivity is consistent with known patterns of flexibility of GNRA tetraloops (28,38). Nucleotides NRA of a GNRA tetraloop are more polymorphic and flexible than the G. The N nucleotide is the most polymorphic of all.
The SHAPE results suggest that the stem-loop replacing helix 91 (Figure 3A) might not form the predicted secondary structure. The stem-loop at this position exhibits a distinctive pattern of reactivity. Two nucleotides in the stem appear to be anomalously flexible. This SHAPE data will inform design changes in future iterations of a-rRNA.
SHAPE characterization of a-rRNA tertiary interactions
RNAs require Mg2+ ions to form compact structures (35-37). The core of the assembled LSU is particularly rich in Mg2+ ions (9,22,39). In our 3D model of the a-PTC, the conformation of the a-rRNA is stabilized in part by Mg2+ ions (Figures 2B and 3B) and also by tertiary interactions between RNA elements that are remote in the primary sequence. Here we ask if a-rRNA in vitro, upon the addition of Mg2+, forms a compact structure consistent with the in silico a-PTC model (Figure 2B).
A network of phosphate-Mg2+-phosphate interactions is anticipated in the a-PTC, based on the network in the LSU crystal structure (PDB 2J01) (22). This phosphate-Mg2+-phosphate network is indicated in Figure 3C, where magenta circles represent nucleotides with phosphate oxygens that make direct contacts with Mg2+ ions (<2.4 Å cutoff distance). Long-range phosphate-Mg2+-phosphate linkages are indicated by magenta lines. The proposed network includes all Mg2+ ions that interact with phosphates of two or more of the nucleotides. Formation of the phosphate-Mg2+-phosphate network is coupled with formation of base-base tertiary interactions. Therefore Mg2+ is expected to influence the rRNA flexibility and SHAPE reactivity of nucleotides that contact Mg2+ or are involved in long-range tertiary interactions. This pattern of Mg2+-dependent SHAPE reactivity has previously been observed for tRNA, RNase P, the P4-P6 domain of the Tetrahymena Group I intron and Domain III of the 23S rRNA (33,40-42).
The influence of Mg2+ on a-rRNA SHAPE reactivity (Figure 3B), suggests that Mg2+ does indeed induce global folding. Some Mg2+-induced changes in SHAPE reactivity of a-rRNA are clustered around hypothesized regions of direct RNA-Mg2+ contact similar to those in the folded LSU. Others are observed in regions of hypothesized long-range tertiary interactions.
Changes in SHAPE reactivity upon addition of Mg2+ (10 mM) were determined. The greatest changes in SHAPE reactivity (>40% change) are mapped onto the predicted secondary structure of a-rRNA (increases: green; decreases: blue, Figure 3B). Nucleotides with large changes in reactivity are dispersed throughout the sequence (Figure S5) and the secondary structure. Nucleotides for which the absolute SHAPE reactivity is small (≤ 0.3) were omitted from the analysis to avoid attributing artificial significance to small changes in absolute value.
Of the 65 rRNA nucleotides (excluding stem-loops) with altered SHAPE reactivities upon addition of Mg2+ (Figure 3B), 25 (38%) are within 3 residues of a nucleotide that contacts an Mg2+ ion in the predicted interaction network in Figure 3C. Most of the other Mg2+ effects are at or near bulges and loops, assumed to be involved in tertiary interactions. The data are consistent with induction by Mg2+ of local and long distance interactions that alter nucleotide flexibility. The results suggest a transition of the a-rRNA from an extended secondary structure to a collapsed structure with tertiary interactions.
The SHAPE reactivities argue against significant alteration of the secondary structure upon addition of Mg2+. Only a small number of helical nucleotides show Mg2+-dependent changes. Of the 65 rRNA nucleotides that show significant Mg2+ effects, only a few (9 nucleotides) are found in regions that are paired in the predicted secondary structure, and several of these are adjacent to predicted bulges or other single-stranded regions.
Magnesium alters the gel mobility of a-rRNA
The SHAPE data support the hypothesis that the conformation of the a-rRNA is altered by Mg2+. This hypothesis is also tested by gel mobility. In the absence of Mg2+, a-rRNA appears to have one predominant conformation and an ensemble of minor conformations (Figure 4), inferred from the smeared intensity between the lower band and fainter upper band on the native gel. The smear does not arise from heterogeneity in length, as a-rRNA runs as a tight single band on denaturing gels. During the course of Mg2+ titration, the a-rRNA is seen in a native gel to transition to a single, slower-running state (Figure 4). This transition appears complete at 250 µM Mg2+.
a-rRNA forms a specific 1:1 complex with a-rPeptide L4 The fluorescence of the tryptophan within a-rPeptide L4 (Table 1) was used to monitor the interaction of a-rRNA and a-rPeptide L4 in a Continuous Variation experiment (43,44). The two species were mixed in a series of solutions of varying mole fractions. The total concentration of the two species was held fixed throughout. Continuous variation of a-rRNA and a-rPeptide L4 shows a distinct inflection at 1:1 stoichiometry (Figure 5), where the molar fractions of a-rPeptide L4 and a-rRNA are both 0.5. Thus continuous variation of a-rRNA and a-rPeptide L4 suggests a 1:1 stoichiometry of binding. In a control experiment, an inflection was not observed for the continuous variation of the P4-P6 domain of the Tetrahymena Group I intron with a-rPeptide L4.
a-rRNA forms complexes with MBP-a-rPeptides L3, L15, and L22
The interactions between a-rRNA and a-rPeptides L2, L3, L15, or L22 are not detectable by fluorescence due to the absence of fluorescent amino acids in these peptides. Instead, the interactions of a-rPeptides L2, L3, L15, or L22 with a-rRNA were evaluated by electrophoretic mobility shift assay (EMSA). The peptides alone are too small to cause a visible change in a-rRNA gel mobility. Therefore the a-rPeptides were fused to the C-terminus of maltose-binding protein (MBP) using molecular cloning techniques. In a two-color EMSA (45), uncomplexed RNA is green, and free protein (MBP-a-rPeptide fusions) is red. If an RNA and a protein form a complex, they co-localize on the gel, producing a yellow band (Figure 6).
The results indicate that MBP fusions with a-rPeptides L3, L15, or L22 form complexes with a-rRNA in the range of 1-10 µM of the MBP-a-rPeptide fusion (Figure 6). Each of these three MBP-a-rPeptide fusions shifts the a-rRNA band on the gel. MBP-a-rPeptide L3 required overnight incubation to form a complex with a-rRNA, but MBP-a-rPeptides L15 and L22 required only a few hours incubation. MBP-a-rPeptide L2 did not form a complex with a-rRNA under any of the conditions tested, even at concentrations an order of magnitude greater than those sufficient for binding of MBP-a-rPeptide L3, L15, or L22. Here we assayed a-rRNA interactions with a-rPeptides in the absence of Mg2+. The a-rRNA exhibits two dominant conformations on EMSA gels (Figure 6) indicated by two bands. The binding of any a-rPeptide, like binding of Mg2+, induces formation of a single state, indicated by a single band on the gel. A control experiment shows MBP alone does not bind to a-rRNA (Figure 6A). At high protein concentration (>50 μM), the a-rRNA is seen to degrade, as indicated by faint low molecular weight RNA bands on the gels.
Using the method of Williamson (46), we have estimated the dissociation constants (Kd) of the complexes formed by MBP-a-rPeptides L3, L15, or L22 with a-rRNA. Data were obtained by integrating the band intensities from the EMSA gels (Figure 6). MBP-a-rPeptide L3 binds weakly, and the Kd is estimated to be greater than 10 µM. The Kd is 4.6 µM for MBP-a-rPeptide L15 and 7.4 µM for MBP-a-rPeptide L22. Kimura previously measured a Kd of 1 µM for the interaction of intact ribosomal protein L2 with appropriate fragments of the 23S rRNA (47). Hachimori measured a Kd of 10 nM for the interaction of intact ribosomal protein L3 with the Sarcin/Ricin Domain of 23S rRNA (48). Nierhaus and others used filter binding and sedimentation to determine stoichiometries of binding of ribosomal proteins to rRNA (49). Ribosomal proteins L2, L3, L4 are classified as normal binders to the 23S rRNA in the presence of 4 mM Mg2+. L15 and L22 are classified as weak binders.
a-rRNA interacts with intact rProteins L3, L4, L15, and L22 in vivo.
To determine if our in vitro results could be replicated in vivo we used the yeast three-hybrid system to investigate interactions of a-rRNA with a-rPeptides and intact rProteins. The interactions of full length rProteins L2, L3, L4, L15, or L22 with a-rRNA were characterized (schematic provided in Figure 7A). Each rProtein was fused to the C terminus of the GAL4 transcriptional activation domain (GAD), which interacts with the LacZ reporter gene. These hybrids are referred to as GAD-L2, GAD-L3, GAD-L4, GAD-L15, and GAD-L22. The RNA-rProtein interactions were assayed by quantification of increased β-gal activity due to enhanced expression of the LacZ reporter gene (Figure 7B). By this assay, a-rRNA exhibited the strongest in vivo interaction with L4, with a β-gal average activity of 1150 Miller Units (MU). The second strongest in vivo interaction with a-rRNA was observed with L22 at 750 MU, and moderate in vivo interactions were observed with L15 (420 MU) and L3 (400 MU). Consistent with the in vitro assays, a-rRNA did not exhibit any measurable in vivo interaction with L2. With a similar experimental approach, interactions were not observed between the a-rPeptides and a-rRNA. Our current hypothesis is that the linker between the a-rPeptide and GAD may need to be lengthened to allow and observe interactions. Further work in this area is in progress.
Translation and the ribosome are some of the most illuminating biochemical links between current and ancient biological systems (50). Translation provides a powerful experimental and theoretical system for exploring life’s oldest biological processes and molecules (2-7). Some parts of the ribosome are thought to predate our current biology of DNA, RNA, and coded protein. Following previous work (10,51-54), we have undertaken an effort to exploit the ribosome as a tool for learning about ancient biology. Ancient components of the LSU, as inferred from consensus among models of early ribosomal evolution, were constructed and characterized. The conclusion here is that the core of the LSU has retained ancient ability to fold and assemble over vast evolutionary time frames of billions of years. Relatively small ancestral components of the LSU are folding-competent and assembly-competent.
The Experimental System. Advances in technology allow us to address fundamental questions of paleobiochemistry, a discipline first conceived by Zuckerkandl and Pauling (55). Here we have constructed a model, in silico, in vitro and in vivo, of an ancestral Peptidyl Transferase Center. The a-PTC is designed from a consensus among previous proposals of ribosomal evolution by the laboratories of Fox (11), Steinberg (8), Noller (12), Williams (9), and others. The a-PTC is an experimental model of one of life’s most ancient assemblies, composed of fragments of rRNA and protein from the oldest part of the extant ribosome (Figure 1). These rRNA fragments are joined together here by stem-loops to form a single RNA polymer called the a-rRNA (Figure 2A). The protein fragments contained within the a-PTC (Table 1) are a-rPeptides (ancestral peptides), derived from portions of rProteins that penetrate deep into the ribosome.
Structure and assembly of the a-PTC was probed by a variety of computational, biophysical and in vivo methods. The results suggest that the a-PTC in vitro and in vivo folds and assembles in general accordance with the predictions of the in silico model. This conclusion is based on (i) RNase H cleavage and SHAPE reactivity, which support formation of the predicted secondary structure of a-rRNA, (ii) the Mg2+-dependence of the SHAPE profile and gel mobility, which indicate LSU-like Mg2+ interactions within a-rRNA, and Mg2+-induced formation of tertiary interactions, and (iii) specific binding of a-rPeptides L3, L4, L15, and L22 (in vitro) and rProteins L3, L4, L15, and L22 (in vivo) with a-rRNA. L2 appears to be an exception in that neither in vitro binding of a-rPeptide L2 nor in vivo binding of rProtein L2 to the a-rRNA is observed. Variants of a-rPeptide L2 will be constructed and assayed for binding in subsequent work.
Yonath and coworkers previously described a model for the proto-ribosome (10,53) that is significantly smaller than model our a-rRNA. In fact, Yonath’s proto-ribosome is fully contained within our a-rRNA. In contrast to the a-PTC, there are no peptide components in the proto-ribosome. Yonath’s proto-ribosome appears to represent a model of a smaller, even more primitive ancestor of the a-PTC.
The Cooption Model of Early Ribosomal Evolution. The fundamental hypothesis that underlies our approach is called the “cooption model” (7,12). In the cooption model, (a) ancestors of the SSU and LSU originated and evolved independently of each other, with autonomous functionalities, (b) an ancestor of the LSU, independent of the SSU, contained the PTC, which catalyzed production of short, non-coded peptide or combined peptide/ester (56) oligomers, (c) some of the non-coded oligomer products of the LSU ancestor associated with the LSU, conferring advantage, (d) an ancestor of the SSU had a function that is more tentative but as proposed by Noller may have involved RNA binding or polymerization, (e) ancestral LSUs and SSUs joined, in a cooption process, enabling coded synthesis, and (f) the peptide/ester oligomers associated with the ancestral LSU eventually became the tails of ribosomal proteins that penetrate deep within the extant LSU.
In the cooption model, and other models of ribosomal evolution, changes over evolution are restricted to those that maintain the structure and functionality of the PTC and the decoding center. The catalytic core of the LSU, and the decoding center of the SSU, are assumed to predate the cooperative relationship between the LSU and SSU. This cooption model predicts that an ancestor of the 23S rRNA, lacking more recent components required for association with the SSU, will retain folding, recognition and assembly capabilities. Here we test that aspect of the cooption model by determining if the a-rRNA, a model of an ancestor of the 23S rRNA, can fold and assemble with proposed ancestral fragments of ribosomal proteins to form the a-PTC.
The results here support elements of the cooption model of ribosomal evolution. The proposed ancestral elements of the LSU are highly robust in folding and assembly. We have shaved around 2400 nucleotides from the 23S rRNA, and the vast majority of amino acids from the protein components, excising globular protein domains in toto. The remaining a-rRNA and a-rPeptides, except that derived from ribosomal protein L2, retain the ability to fold and specifically assemble. It is striking that the a-rPeptides, lacking α-helices, β-sheets, and globular domains, retain the ability to interact specifically with the a-rRNA. This robustness is consistent with the premise that the ribosome is an ancient assembly that evolved in a primitive biological environment and has survived billions of years of evolution, growing in size and complexity, without major changes in core structure or function. Although the modern LSU has the appearance of a massive monolithic assembly (23), our results here, and elsewhere (41), indicate that it can be understood as much smaller elements of RNA and peptide that retain ancient abilities to fold and independently assemble.
References
1. Mears, J.A., Cannone, J.J., Stagg, S.M., Gutell, R.R., Agrawal, R.K. and Harvey, S.C. (2002) Modeling a Minimal Ribosome Based on Comparative Sequence Analysis. J. Mol. Biol., 321, 215-234.
2. Woese, C.R. (2000) Interpreting the Universal Phylogenetic Tree. Proc. Natl. Acad. Sci. U. S. A., 97, 8392-8396.
3. Woese, C.R. (2001) Translation: In Retrospect and Prospect. RNA, 7, 1055-1067.
4. Fournier, G.P., Neumann, J.E. and Gogarten, J.P. (2010) Inferring the Ancient History of the Translation Machinery and Genetic Code Via Recapitulation of Ribosomal Subunit Assembly Orders. PLoS One, 5, e9437.
5. Williams, D., Fournier, G.P., Lapierre, P., Swithers, K.S., Green, A.G., Andam, C.P. and Gogarten, J.P. (2011) A Rooted Net of Life. Biol. Direct, 6, 45.
6. Wolf, Y.I. and Koonin, E.V. (2007) On the Origin of the Translation System and the Genetic Code in the RNA World by Means of Natural Selection, Exaptation, and Subfunctionalization. Biol. Direct, 2, 14.
7. Fox, G.E. (2010) Origin and Evolution of the Ribosome. Cold Spring Harb. Perspect. Biol., 2, a003483.
8. Bokov, K. and Steinberg, S.V. (2009) A Hierarchical Model for Evolution of 23S Ribosomal RNA. Nature, 457, 977-980.
9. Hsiao, C., Mohan, S., Kalahar, B.K. and Williams, L.D. (2009) Peeling the Onion: Ribosomes Are Ancient Molecular Fossils. Mol. Biol. Evol., 26, 2415-2425.
10. Belousoff, M.J., Davidovich, C., Zimmerman, E., Caspi, Y., Wekselman, I., Rozenszajn, L., Shapira, T., Sade-Falk, O., Taha, L., Bashan, A. et al. (2010) Ancient Machinery Embedded in the Contemporary Ribosome. Biochem. Soc. Trans., 38, 422-427.
11. Hury, J., Nagaswamy, U., Larios-Sanz, M. and Fox, G.E. (2006) Ribosome Origins: The Relative Age of 23S rRNA Domains. Orig Life Evol Biosph, 36, 421-429.
12. Noller, H.F. (2010) Evolution of Protein Synthesis from an RNA World. Cold Spring Harb. Perspect. Biol., 7, 7.
13. Rich, A. (1962) In Kasha, M. and Pullman, B. (eds.), Horizons in Biochemistry. New York: Academic, pp. 103–126.
14. Woese, C.R. (1967) The Genetic Code: The Molecular Basis for Genetic Expression. Harper & Row, N.Y.
15. Gilbert, W. (1986) Origin of Life: The RNA World. Nature, 319, 618-618.
16. Crick, F.H. (1968) The Origin of the Genetic Code. J. Mol. Biol., 38, 367-379.
17. Orgel, L.E. (1968) Evolution of the Genetic Apparatus. J. Mol. Biol., 38, 381-393.
18. Crick, F. (1970) Central Dogma of Molecular Biology. Nature, 226, 561-563.
19. Rost, B. (1997) Protein Structures Sustain Evolutionary Drift. Fold Des., 2, S19-24.
20. Heinz, D.W., Baase, W.A., Zhang, X.J., Blaber, M., Dahlquist, F.W. and Matthews, B.W. (1994) Accommodation of Amino Acid Insertions in an Alpha-Helix of T4 Lysozyme. Structural and Thermodynamic Analysis. J. Mol. Biol., 236, 869-886.
21. Selmer, M., Dunham, C.M., Murphy, F.V., Weixlbaumer, A., Petry, S., Kelley, A.C., Weir, J.R. and Ramakrishnan, V. (2006) Structure of the 70S Ribosome Complexed with mRNA and tRNA. Science (New York, N.Y.), 313, 1935-1942.
22. Hsiao, C. and Williams, L.D. (2009) A Recurrent Magnesium-Binding Motif Provides a Framework for the Ribosomal Peptidyl Transferase Center. Nucleic Acids Res., 37, 3134-3142.
23. Ban, N., Nissen, P., Hansen, J., Moore, P.B. and Steitz, T.A. (2000) The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 Å Resolution. Science (New York, N.Y.), 289, 905-920.
24. Noller, H.F., Hoffarth, V. and Zimniak, L. (1992) Unusual Resistance of Peptidyl Transferase to Protein Extraction Procedures. Science (New York, N.Y.), 256, 1416-1419.
25. Klein, D.J., Moore, P.B. and Steitz, T.A. (2004) The Roles of Ribosomal Proteins in the Structure Assembly, and Evolution of the Large Ribosomal Subunit. J. Mol. Biol., 340, 141-177.
26. Ben-Shem, A., Jenner, L., Yusupova, G. and Yusupov, M. (2010) Crystal Structure of the Eukaryotic Ribosome. Science (New York, N.Y.), 330, 1203-1209.
27. Petrov, A., Bernier, C., Hsiao, C., Okafor, C.D., Tannenbaum, E., Stern, J., Gaucher, E., Schneider, D.M., Harvey, S.C. and Williams, L.D. (2012) RNA-Magnesium-Protein Interactions in Large Ribosomal Subunit. J. Phys. Chem. B, 116, 8113-8120.
28. Mohan, S., Hsiao, C., Bowman, J.C., Wartell, R. and Williams, L.D. (2010) RNA Tetraloop Folding Reveals Tension between Backbone Restraints and Molecular Interactions. J. Am. Chem. Soc., 132, 12679-12689.
29. Kauffmann, A.D., Campagna, R.J., Bartels, C.B. and Childs-Disney, J.L. (2009) Improvement of RNA Secondary Structure Prediction Using Rnase H Cleavage and Randomized Oligonucleotides. Nucleic Acids Res., 37, e121.
30. Donis-Keller, H. (1979) Site Specific Enzymatic Cleavage of RNA. Nucleic Acids Res., 7, 179-192.
31. Hofacker, I.L., Fekete, M., Flamm, C., Huynen, M.A., Rauscher, S., Stolorz, P.E. and Stadler, P.F. (1998) Automatic Detection of Conserved RNA Structure Elements in Complete RNA Virus Genomes. Nucleic Acids Res., 26, 3825-3836.
32. Wu, P., Nakano, S. and Sugimoto, N. (2002) Temperature Dependence of Thermodynamic Properties for DNA/DNA and RNA/DNA Duplex Formation. Eur. J. Biochem., 269, 2821-2830.
33. Merino, E.J., Wilkinson, K.A., Coughlan, J.L. and Weeks, K.M. (2005) RNA Structure Analysis at Single Nucleotide Resolution by Selective 2’-Hydroxyl Acylation and Primer Extension (SHAPE). J. Am. Chem. Soc., 127, 4223-4231.
34. Deigan, K.E., Li, T.W., Mathews, D.H. and Weeks, K.M. (2009) Accurate SHAPE-Directed RNA Structure Determination. Proc. Natl. Acad. Sci. U. S. A., 106, 97-102.
35. Brion, P. and Westhof, E. (1997) Hierarchy and Dynamics of RNA Folding. Annu. Rev. Biophys. Biomol. Struct., 26, 113-137.
36. Tinoco, I., Jr. and Bustamante, C. (1999) How RNA Folds. J. Mol. Biol., 293, 271-281.
37. Bowman, J.C., Lenz, T.K., Hud, N.V. and Williams, L.D. (2012) Cations in Charge: Magnesium Ions in RNA Folding and Catalysis Curr. Opin. Struct. Biol., 22, 262-272.
38. Butcher, S.E., Dieckmann, T. and Feigon, J. (1997) Solution Structure of a GAAA Tetraloop Receptor RNA. EMBO J., 16, 7490-7499.
39. Klein, D.J., Moore, P.B. and Steitz, T.A. (2004) The Contribution of Metal Ions to the Structural Stability of the Large Ribosomal Subunit. RNA, 10, 1366-1379.
40. Mortimer, S.A. and Weeks, K.M. (2008) Time-Resolved RNA SHAPE Chemistry. J. Am. Chem. Soc., 130, 16178-16180.
41. Athavale, S.S., Gossett, J.J., Hsiao, C., Bowman, J.C., O’Neill, E., Hershkovitz, E., Preeprem, T., Hud, N.V., Wartell, R.M., Harvey, S.C. et al. (2012) Domain III of the T. thermophilus 23S rRNA Folds Independently to a near-Native State. RNA, 18, 752-758.
42. Athavale, S.S., Petrov, A.S., Hsiao, C., Watkins, D., Prickett, C.D., Gossett, J.J., Lie, L., Bowman, J.C., O’Neill, E., Bernier, C.R. et al. (2012) RNA Folding and Catalysis Mediated by Iron (II). PLoS ONE, 7, e38024.
43. Job, P. (1928) Studies on the Formation of Complex Minerals in Solution and on Their Stability. Annales De Chimie France, 9, 113-203.
44. Cantor, C. and Schimmel, P. (1984) Biophysical Chemistry (I-III). Academic Press, New York.
45. Shcherbakov, D. and Piendl, W. (2007) A Novel View of Gel-Shifts: Analysis of RNA-Protein Complexes Using a Two-Color Fluorescence Dye Procedure. Electrophoresis, 28, 749-755.
46. Van Camp, G., Chapelle, S. and De Wachter, R. (1993) Amplification and Sequencing of Variable Regions in Bacterial 23S Ribosomal RNA Genes with Conserved Primer Sequences. Curr. Microbiol., 27, 147-151.
47. Williams, L.D. (2012) Year of the Solar System: Origin of Life Talk.
48. Barkholt, V. and Jensen, A.L. (1989) Amino Acid Analysis: Determination of Cysteine Plus Half-Cystine in Proteins after Hydrochloric Acid Hydrolysis with a Disulfide Compound as Additive. Anal. Biochem., 177, 318-322.
49. Marquardt, O., Roth, H.E., Wystup, G. and Nierhaus, K.H. (1979) Binding of Escherichia Coli Ribosomal Proteins to 23S RNA under Reconstitution Conditions for the 50S Subunit. Nucleic Acids Res., 6, 3641-3650.
50. Cech, T.R. (2009) Crawling out of the RNA World. Cell, 136, 599-602.
51. Anderson, R.M., Kwon, M. and Strobel, S.A. (2007) Toward Ribosomal RNA Catalytic Activity in the Absence of Protein. J Mol Evol., 64, 472-483.
52. Khaitovich, P., Mankin, A.S., Green, R., Lancaster, L. and Noller, H.F. (1999) Characterization of Functionally Active Subribosomal Particles from Thermus Aquaticus. Proc. Natl. Acad. Sci. U. S. A.. 96, 85-90.
53. Krupkin, M., Matzov, D., Tang, H., Metz, M., Kalaora, R., Belousoff, M.J., Zimmerman, E., Bashan, A. and Yonath, A. (2011) A Vestige of a Prebiotic Bonding Machine Is Functioning within the Contemporary Ribosome. Philos Trans R Soc Lond B Biol Sci, 366, 2972-2978.
54. Ostergaard, P., Phan, H., Johansen, L.B., Egebjerg, J., Ostergaard, L., Porse, B.T. and Garrett, R.A. (1998) Assembly of Proteins and 5S rRNA to Transcripts of the Major Structural Domains of 23S rRNA. J. Mol. Biol., 284, 227-240.
55. Pauling, L. and Zuckerkandl, E. (1963) Chemical Paleogenetics Molecular Restoration Studies of Extinct Forms of Life. Acta Chem. Scand., 17, 9-16.
56. Rich, A. (1971) In Buvet, R. and Ponnamperuma, C. (eds.), Chemical Evoltuion and the Origin of Life. North-Holland Publishing Company, Amsterdam.
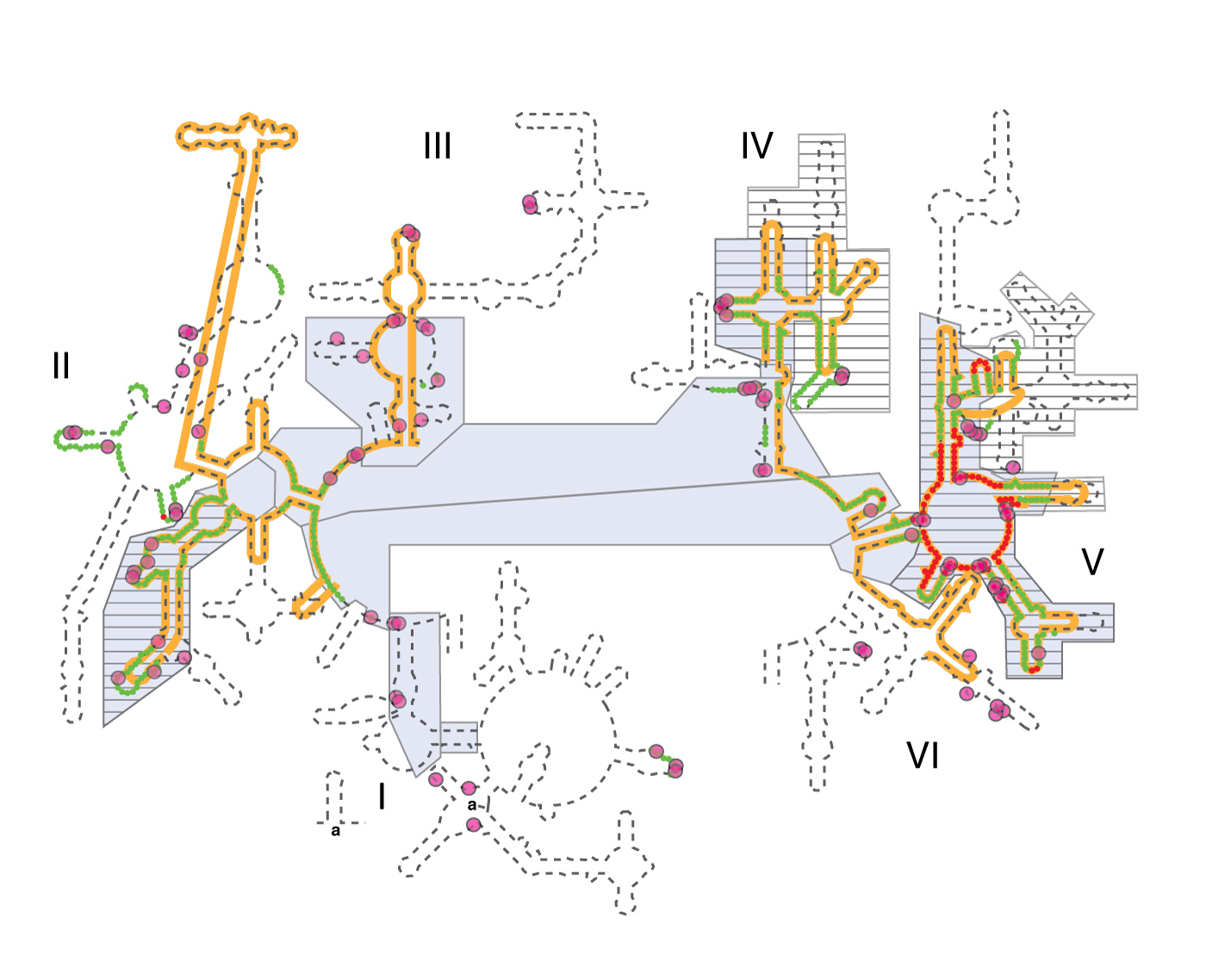
Various models of 23S rRNA evolution. The dashed line illustrates the canonical secondary structure of the T. thermophilus 23S rRNA. Secondary structural domains are indicated by Roman numerals. The red and green lines show the two inner shells of the ribosomal onion of Hsiao and Williams, marking the rRNA that is in closest proximity, in three dimensions, to the site of peptidyl transfer. The gray boxes are ancient according to the ‘A-minor’ method of Steinberg. The hashed boxes (with black horizontal lines) are ancient according to the networking analysis of Fox. Multidentate Mg2+-phosphate interactions, also proposed as an indicator of ancient rRNA, are indicated by magenta circles. The orange line shows the universally conserved portions of the 23S rRNA in bacteria, archaea, eukarya, and in mitochondria, as determined by Gutell and Harvey.
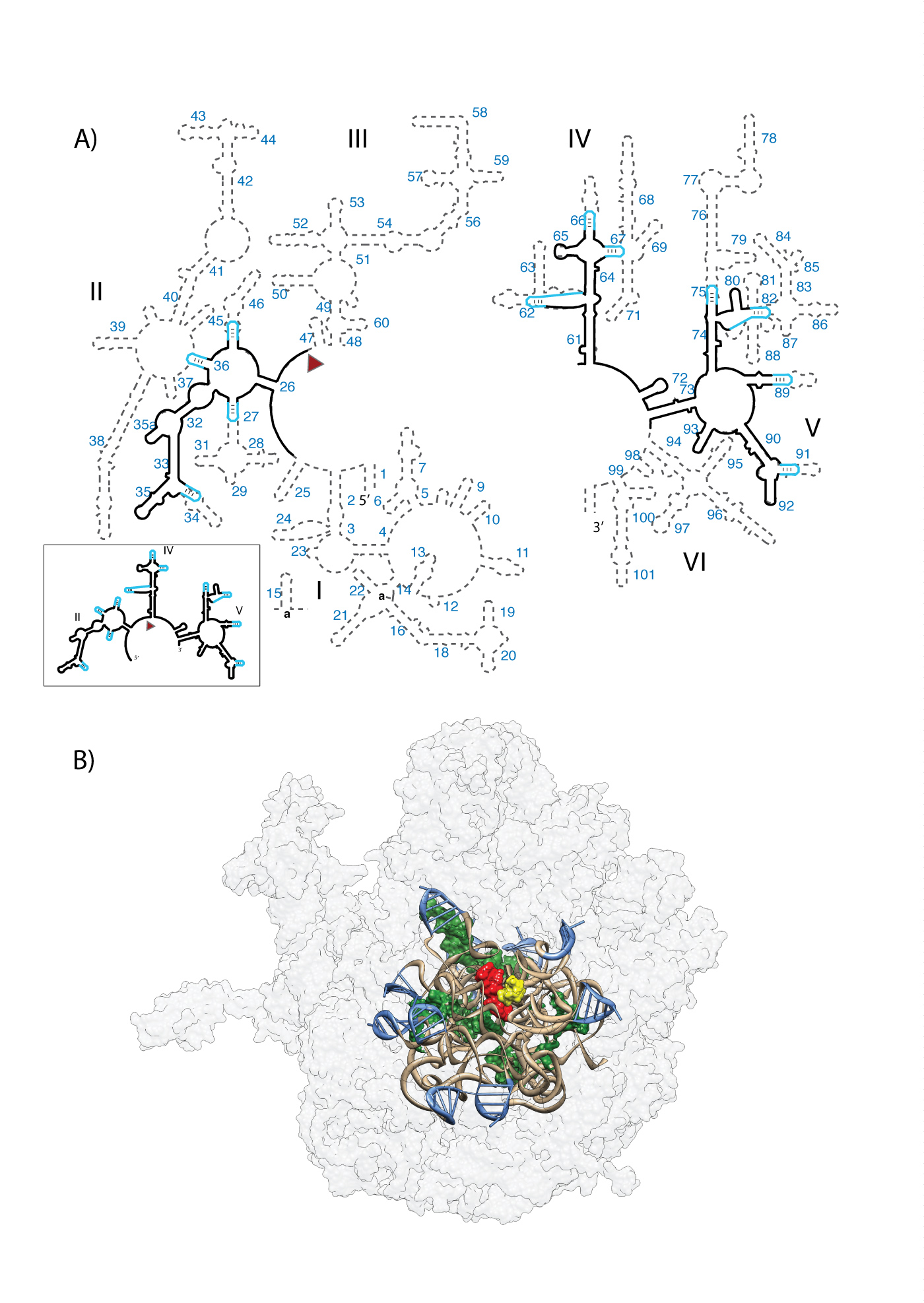
A) Predicted secondary structure of the ancestral rRNA. Ancestral fragments of rRNA, indicated by black lines in the secondary structure, are derived from a consensus of models of rRNA evolution. The ancestral rRNA elements are stitched together by stem-loops (blue). The RNA sequences are from the T. thermophilus 23S rRNA. Helix numbers are indicated. The predicted secondary structure of the a-rRNA alone is highlighted in the outbox. (B) Three-dimensional model of the a-PTC. This 3D model contains the a-rRNA plus five a-rPeptides (ancestral fragments of ribosomal proteins L2, L3, L4, L15, and L22). a-rRNA is shown in ribbon (brown), the stem-loops are blue, and the peptides are surface representation (green). For reference, A-site (yellow) and P-site (red) substrates are shown in the figure, but are not components of the a-PTC. The modern LSU surface is shown for comparison (light gray, transparent).
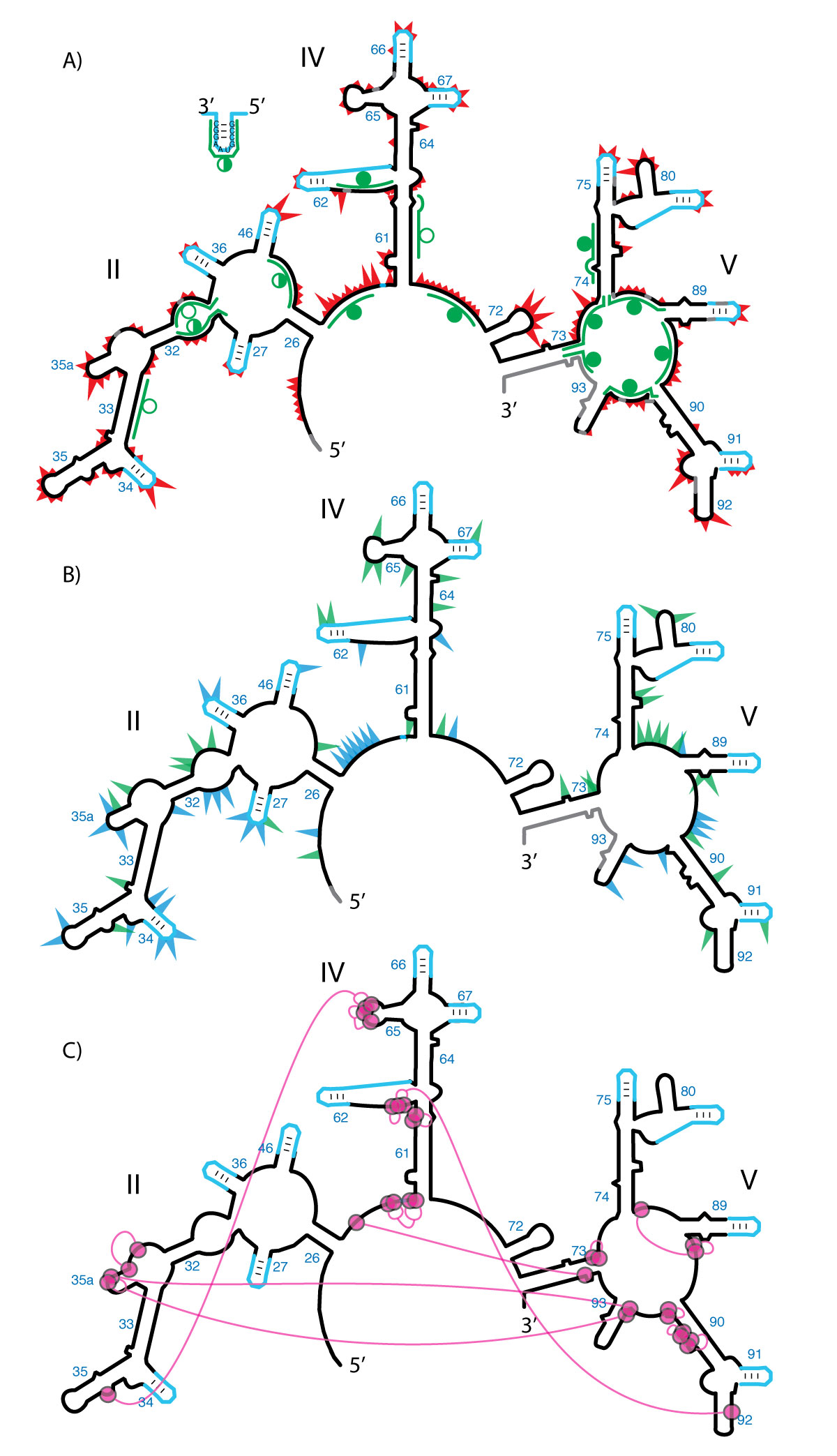
Probing the secondary and tertiary structure of a-rRNA. A) SHAPE and RNase H mapping. Red triangles mark SHAPE reactivities in 250 mM Na+, mapped onto the predicted secondary structure of a-rRNA. Larger triangles indicate greater SHAPE reactivity. RNase H DNA probes are indicated by green lines. Circles indicate extent of RNA digestion by RNase H: filled circles (more than 75%), half-filled circles (between 25% and 75%), and empty circles (less than 25%). B) Effects of 10 mM Mg2+ on SHAPE reactivity suggest formation of tertiary structure. Green triangles show the greatest increases in SHAPE reactivity upon addition of Mg2+. Blue triangles show the greatest decreases in reactivity. C) Multidentate Mg2+-phosphate interactions observed in the T. thermophilus LSU (PDB 2J01) are mapped onto the predicted secondary structure of a-rRNA. Magenta circles indicate first-shell Mg2+-OP (magnesium-phosphate oxygen) interactions. Magenta lines indicate PO-Mg2+-OP linkages. Gray shading in panels A and B indicates rRNA where SHAPE data were not accessible. SHAPE reactions were performed in 50 mM NaHEPES pH 8.0, 200 mM NaOAc, 0 or 10 mM MgCl2.
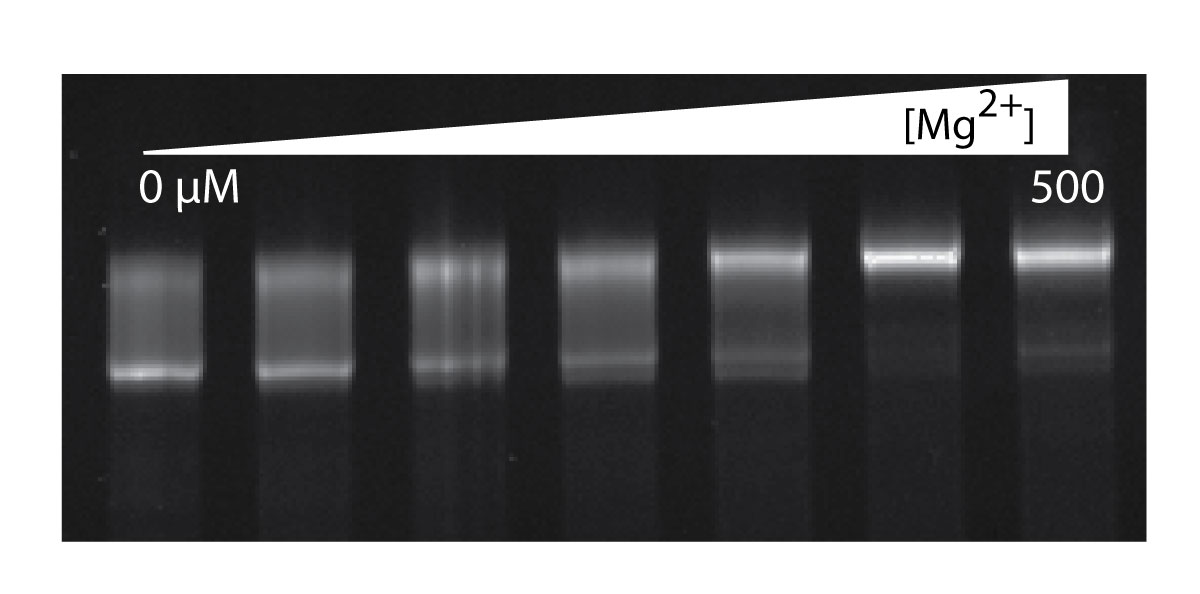
The effect of Mg2+ on gel mobility of the a-rRNA suggests Mg2+ induction of folding. Shown here is a-rRNA annealed in 10 mM Tris, pH 8.0, and varying [Mg2+], resolved on a 5% native acrylamide gel. Lane 1, [Mg2+] = 0 μM; Lane 2, 12.5; Lane 3, 25; Lane 4, 50; Lane 5, 100; Lane 6, 250; Lane 7, 500.
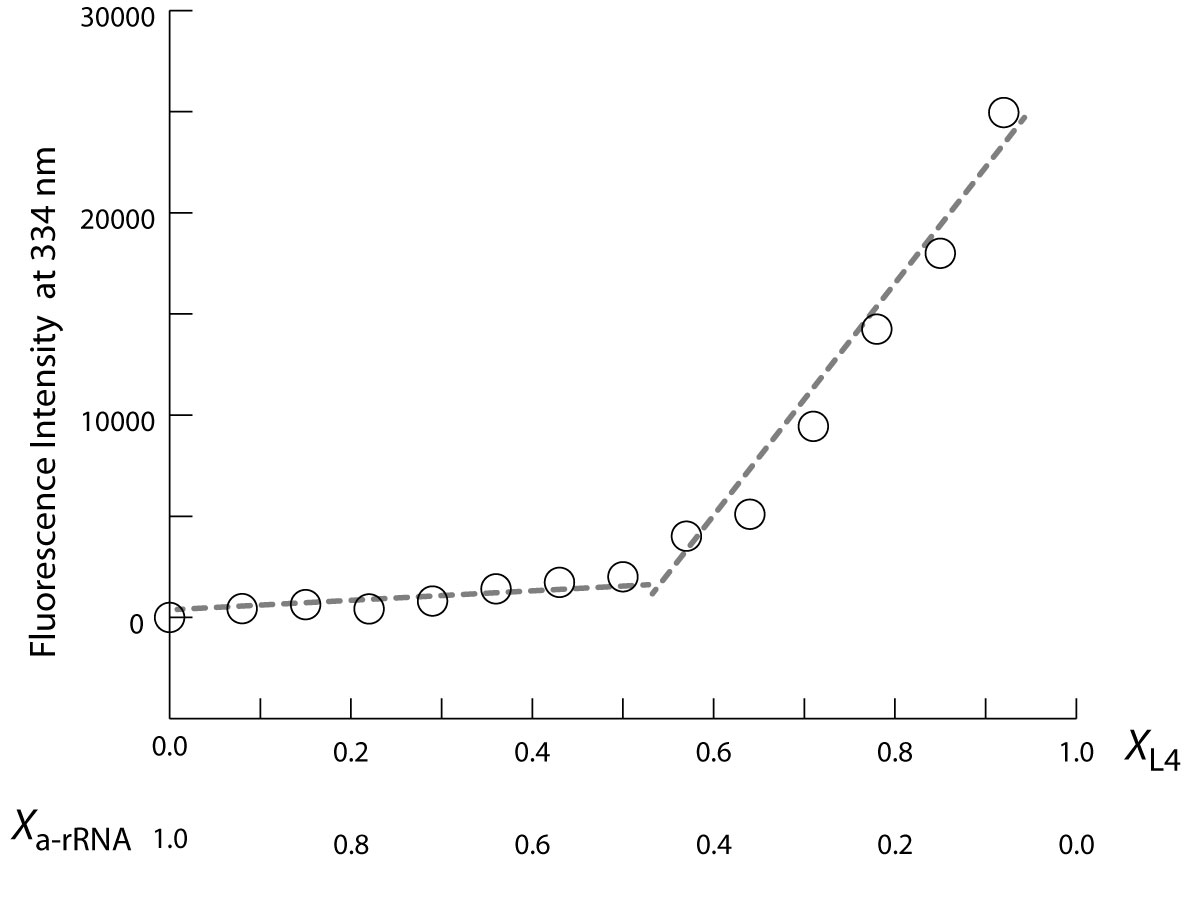
a-rRNA and a-rPeptide L4 are assembly competent and form a complex with 1:1 stoichiometry. Fluorescence signal at 334 nm (F334) is from the tryptophan residue of the a-rPeptide L4. In this plot of a continuous variation experiment of a-rPeptide L4 with a-rRNA, XL4 and Xa-rRNA values on horizontal axis denote mole fraction of the peptide and RNA in each sample, where the total concentration ([a-rRNA] + [a-rPeptide L4]) was held constant at 60 µM. The discontinuity at equivalent mole fractions of peptide and RNA indicates a complex with 1:1 stoichiometry. These binding assays were performed in 10 mM Tris, pH 8.0 at 25 °C.
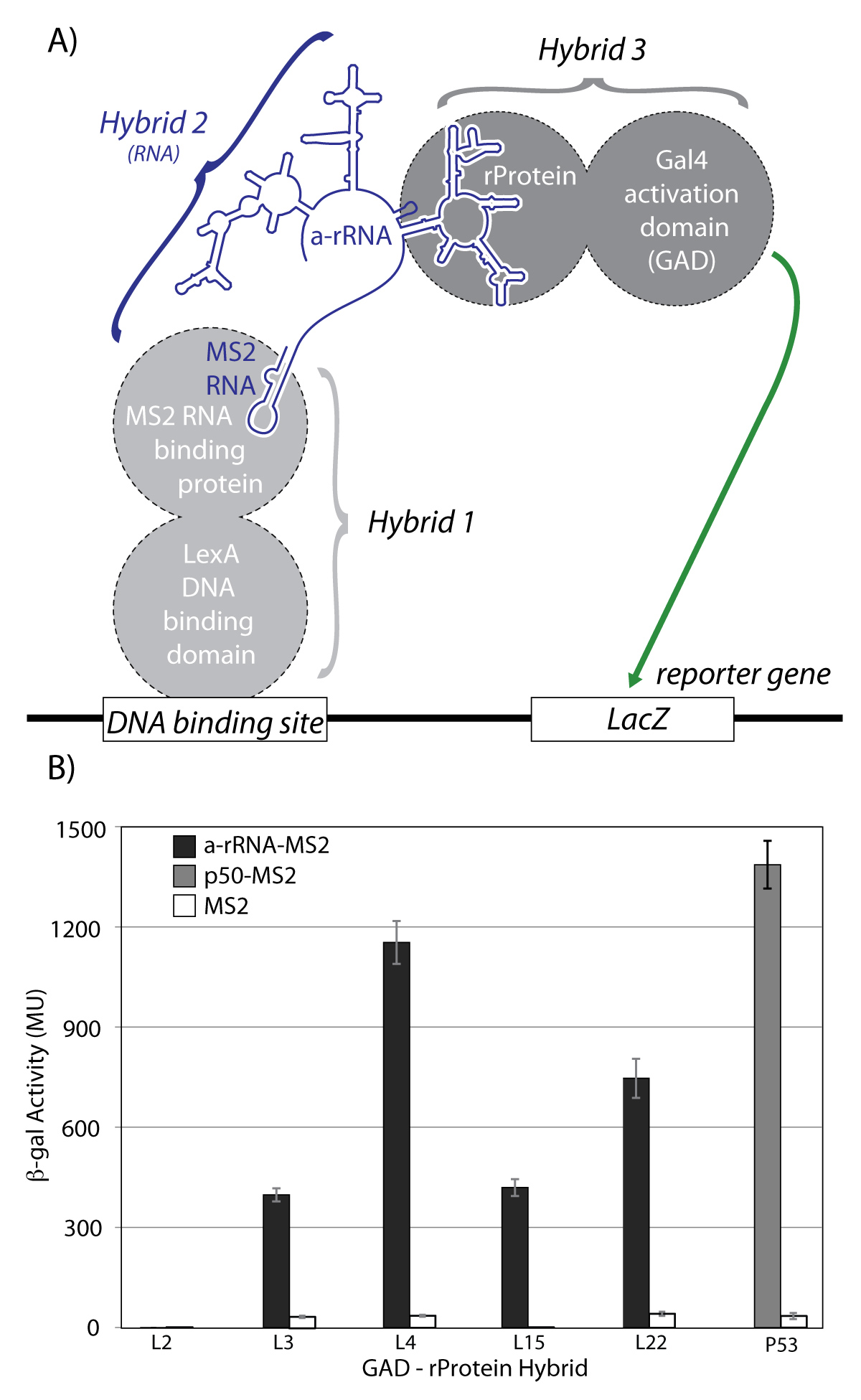
A) Schematic of the yeast three-hybrid assay used to characterize interactions between a-rRNA and ribosomal proteins. In yeast strain YBZ-1, the LacZ reporter gene is controlled by the LexA operator. Hybrid 1, a LexA/MS2 binding protein fusion, binds to the DNA binding site. The MS2 coat protein domain binds tightly to the MS2 RNA, which is fused to the RNA sequence of interest (e.g. a-rRNA). The rProtein of interest is fused to the yeast GAL4 transcriptional activation domain (GAD). In vivo RNA-protein binding results in expression of the LacZ reporter gene, which is quantified by β-galactosidase activity. B) in vivo interactions between a-rRNA and individual rProteins. Interaction is quantified by β-galactosidase activity, reported in Miller Units (MU). Interactions were assayed between a-rRNA-MS2 and GAD-L2, GAD-L3, GAD-L4, GAD-L15, and GAD-L22 (black bars). Positive control was RNA aptamer p50-MS2 and GAD-p53 (grey bar). Negative controls consisted of MS2 RNA and the indicated protein hybrid (white bars).
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Shreyas Athavale
Postdoc
Dana Cook-Schneider
Postdoc
Chiaolong Hsiao
Postdoc
Emmanuel Tannenbaum
Postdoc
Madhan Tirumalai
Postdoc
Eric Anderson
Research Staff
Jessica Bowman
Research Staff
Eric O'Neill
Research Staff
Josh Canzoneri
Graduate Student
Po-Yu Fang
Graduate Student
Jared Gossett
Graduate Student
Timothy Lenz
Graduate Student
Lively Lie
Graduate Student
Thanawadee Preeprem
Graduate Student
Quyen Tran
Graduate Student
Arren Washington
Graduate Student
Amy Boudreau
Undergraduate Student
Kaylee Goss
Undergraduate Student
Suk Hahm
Undergraduate Student
Jason Murray
Undergraduate Student
Jessica Peters
Undergraduate Student
Joshua Raimist
Undergraduate Student
Catherine Trippe
Undergraduate Student
Andrew Warren
Undergraduate Student
-
RELATED OBJECTIVES:
Objective 3.2
Origins and evolution of functional biomolecules
Objective 4.2
Production of complex life.



