2010 Annual Science Report
 Montana State University
Reporting | SEP 2009 – AUG 2010
Montana State University
Reporting | SEP 2009 – AUG 2010
Minerals to Enzymes: The Path to CO Dehydrogenase/Acetyl – CoA Synthase
Project Summary
The relationship between structure and reactivity of iron-sulfur minerals and the active sites of iron-sulfur enzymes is too strong to be coincidental. We and others have proposed that the emergence and genesis of iron-sulfur cluster enzymes occurred by a stepwise process in which mineral motifs were first nested in simple organic polymers and then in response to selective pressure evolved specific gene encoded protein nest that confer high specific enzyme activities. We are examining this hypothesis through nesting NiFeS motifs in a variety of organic nest and examining the structural determinants of reactivity.
Project Progress
We and others have proposed that the emergence and genesis of iron-sulfur enzymes occurred in a stepwise process whereby iron-sulfur mineral motifs or iron-sulfur clusters were first nested in simple organic polymers such as short peptides and that there evolutionary path either involved 1) refinement of the nest in response to selective pressure to increase catalytic function and/or 2) advancing reactivity by adopting superior nests from existing proteins having other functions. The project involves examining these possible evolutionary transitions from abiotic to biological catalysts using a two pronged approach involving 1) a more biochemical approach using known protein nests as a template for the incorporation of hetermetalic metal clusters and 2) a more inorganic synthetic chemistry approach of directed synthesis of mimetic clusters with small molecule or small peptide ligands. The unifying features of the project are that the products of both synthetic strategies are interrogated with respect to their metal cluster composition, metal coordination, and CO oxidation reactivity.
In the more biochemical thrust of the project we have been reconstituting empty nest of known metalloenzymes which we know from previous characterization pocess empty cavities in the absence of their native metal sites with appropriate ligands to bind metals and/or metal clusters. For these studies we have been using the native peptide two proteins 1) an [FeFe]-hydrogenase and 2) the radical SAM enzyme HydE. We have obtained very encouraging preliminary results from the studies involving using the [FeFe]-hydrogenase structural protein. We have shown that if the native active site metal cluster of [FeFe]-hydrogenase is removed and the metal free structural protein is subsequently incubated with a mixture of Ni, Fe, and S a reactive CO oxidation catalyst can be generated. We are currently optimizing the conditions of the experiment to enable us to determine the result structure of the NiFeS active site and examine the key determinants for catalysis.
In parallel, we are exploring a new synthetic strategy to define a template for synthesizing heterometal substituted [M-3Fe-4S] clusters in gram quantities that are related to the catalytic active clusters of nitrogenase FeMo-co and carbon monoxide dehydrogenase Ni-containing C-cluster. Using dithiocarbamate ligands, we have synthesized the 'parent’ compound. We are exploring the synthetic pathways of selectively derivatizing the coordination environment of the heterometal vs. the iron sites.
Using chelating sulfur ligands we are approaching the synthesis of [M-3Fe-4S] clusters from an alternative route that also allow us to obtain mimetic compounds for SAM radical enzymes that catalyze a large group of radical-based transformations in biochemistry. Using a tridentate ligand the critical 3:1 site differentiation of a [4Fe-4S] cluster can be achieved, which allows for demetallation as well as selective derivatization of the unique Fe site. We have synthesized the organic LS3 ligand and the demetallation and derivatization steps are in progress.
Lastly, the close to atomic resolution (1.4 Å) structure of the C-cluster was analyzed by mapping out the immediate protein environment in order to define a minimal amino acid sequence that may provide nesting for a [Ni-4Fe-4S] cluster. The synthetic efforts described above provide the synthetic methodology to attempt the actual synthesis of the nested cluster in a minimal protein environment. Analysis of the crystal structure (PDB ID: 3B53) revealed that the C-cluster is partially exposed to aqueous solvent environment. The cluster is covalently coordinated by five cysteine thiolate and a histidine imidazole rings. Interestingly the first coordination sphere defined by 3.75 Å environment is limited to a few C-H…S, O-H…S hydrogen bonds, a dipole interaction with Gly backbone, and an ionic interactions from a nearby lysine residue. An additional 3.75 Å environment now reveals a much larger array of weak interactions, however; due to the considerable distance (~6 Å or more) from the central cluster their importance in affecting catalysis will likely be limited. Thus a minimalist amino acid sequence that has potential for nesting a [Ni-4Fe-4S] cluster is GxHG>3)CG(>3)CG(>3)CG(>3)CG(>3)CGx with strategically placed glutamate and lysine residues in the 2nd and 6th groups of glycine residues relative to the histidine position.
![Structure of the 2Fe Subcluster Deficient State of an Algal [FeFe]-Hydrogenase](../../../../media/site-content/reports/2010/mon/w3dlhyvmkc_hydrogenase-copy.jpg)
The [FeFe]-hydrogenase in the 2Fe subcluster deficient state exists with an open channel leading to a central cavity with protein thiolate ligands (cysteine residues) poised for metal and metal cluster binding. This form of the hydrogenase has been used for nesting, cluster binding and reactivity experiments to examine if heterometallic sites (NiFe) can be built on this scaffolding and can confer CO oxidation reactivity. a. Surface representation of 2Fe subcluster deficient state of an [FeFe]-Hydrogenase (blue) superimposed on native [FeFe]-hydrogenase (green ribbon representation). b, The 2Fe subcluster deficient state of an [FeFe]-Hydrogenase exists with a channel leading to the active site 2Fe subcluster recipient cavity. c, d, Electrostatic surface representations of the 2Fe subcluster deficient state of an [FeFe]-Hydrogenase and the native state, respectively, calculated with the PyMol plugin APBS. Negative (-10 kT) in red and positive (10 kT) in blue.
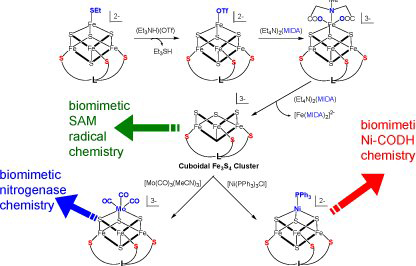
Synthetic strategy for creating a library of heterometal substituted molecular Fe-S clusters (adopted from Holm R.H. Pure and Appl. Chem. 1998, 70, 931)
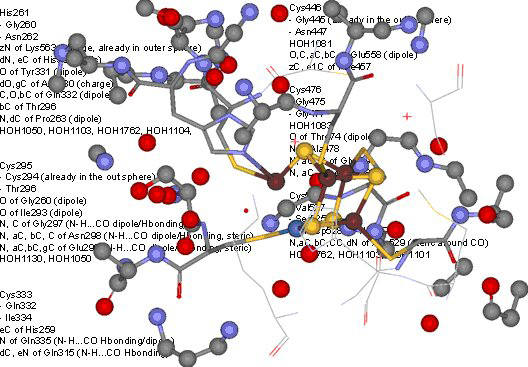
Network of weak interactions within 3.75Å of the C-cluster of NiCODH defining a minimal protein binding motif for nesting experiments of Ni-Fe-S clusters
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Robert Szilagyi
Project Investigator
Joan Broderick
Co-Investigator
James Ferry
Co-Investigator
John Peters
Co-Investigator
Michael Russell
Co-Investigator
Martin Schoonen
Co-Investigator
Guana Siluvai Pitchai
Postdoc
Logan Giles
Doctoral Student
Travis Harris
Doctoral Student
Kevin Swanson
Doctoral Student
Mingyu Wang
Doctoral Student
-
RELATED OBJECTIVES:
Objective 3.1
Sources of prebiotic materials and catalysts
Objective 3.2
Origins and evolution of functional biomolecules
Objective 3.3
Origins of energy transduction
Objective 3.4
Origins of cellularity and protobiological systems
Objective 7.1
Biosignatures to be sought in Solar System materials
Objective 7.2
Biosignatures to be sought in nearby planetary systems