2009 Annual Science Report
 University of Hawaii, Manoa
Reporting | JUL 2008 – AUG 2009
University of Hawaii, Manoa
Reporting | JUL 2008 – AUG 2009
Microbial Ecology in Hawaiian Lava Caves
Project Summary
We have been studying a microbial biofilm growing at very low light intensities and high temperature and humidity below the entrance of a lava cave in Kilauea Crater, Hawai’i Volcanoes National Park. The cave presents an oligotrophic environment, but condensation of geothermally heated groundwater that vents at the rear of the cave has promoted the development of a complex microbial community, similar in higher order taxonomic structure to copiotrophic soil environments. Given the existence of lava tubes of similar geologic composition on Mars, geothermal activity there may have allowed the existence, or persistence, of complex microbial communities in similar Martian environments, wherein they would be shielded from the effects of harmful UV radiation.
Project Progress
We have continued to analyze metagenomic data (randomly sequenced DNA fragments) and Bacterial V6-ribosomal tag sequence (mediated by massively parallel 454 sequencing technology) to provide a comprehensive taxonomic inventory of the mat bacterial community. Both these analyses were conducted from a single sample of DNA extracted from the purple-pigmented microbial mat.
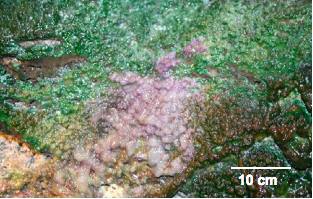
Figure 1. Luxuriant microbial growth, consisting of purple and green portions, is observed on the cave roof in the twilight zone near the entrace to the cave, supported by condensation of geothermally heated ground water.
In total, 386,217 random DNA fragment reads (made up by 95,386,202 bases) were returned from 454 Life Sciences Corporation. Newbler Assembler software v 1.1.03.24 was used to assemble the reads into 37,143 contigs (14,163,791 bases). The prokaryotic gene-finding program MetaGene was applied to predict genes in the assembled contigs and 43,712 genes were predicted. Singletons and genes predicted by MetaGene were compared by BLAST (e-value cutoff of 10-7) to a database containing all protein-coding genes (4,229,793 sequences from 962 species) in KEGG (ftp://ftp.genome.jp/pub/kegg/genes/fasta/genes.pep). The domain of origin from the best high scoring pair (HSP) of each query sequence was then determined. On the basis of the species predicted according to BLAST, closely related genomes were manually selected as the query database for analysis using KAAS (KEGG Automatic Annotation Server, http://www.genome.jp/tools/kaas/) as the accuracy of KAAS is improved if closely related species of the query are contained in the dataset. Orthologous genes were computationally deduced using the bi-directional best hit (BBH) method and KEGG orthology (KO) identifiers were assigned to these genes. This enabled the reconstruction of KEGG pathways and BRITE hierarchies and the network of pathways present within the mat community are illustrated in Figure 2.

Figure 2. The network map depicting metabolic pathways occuring in the microbial mat community. Red, yellow and blue lines represent genes present in the sample whereas white lines depicts genes that are not present.
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Stuart Donachie
Project Investigator
Mark Brown
Collaborator
Gayle Philip
Postdoc
-
RELATED OBJECTIVES:
Objective 5.1
Environment-dependent, molecular evolution in microorganisms
Objective 5.3
Biochemical adaptation to extreme environments