2009 Annual Science Report
 University of Hawaii, Manoa
Reporting | JUL 2008 – AUG 2009
University of Hawaii, Manoa
Reporting | JUL 2008 – AUG 2009
Mechanisms of Marine Microbial Community Structuring
Project Summary
The sub-tropical open ocean is an extreme environment that presents the opportunity to examine the factors affecting microbial community structure across a number of environmental gradients. We have developed and utilized a novel assay that allows us to simultaneously determine the taxonomic composition of Archaea, Bacteria and microbial Eucarya in DNA extracted from environmental samples. Samples analyzed represent the epi-, meso- and bathy-pelagic zones of the ocean, which display gradients in temperature, pressure, oxygen content, nutrient content and photosynthetically available radiation.
Project Progress
We concluded the ribosomal tag pyrosequencing study of the phylogenetic diversity of Archaea, Bacteria and Eucarya over a depth profile at the Hawaii Ocean Time-Series Station, ALOHA and published it in the ISME Journal.
This work involved amplifying the V9 region of the SSU rRNA gene from samples representing the epi- (10 m), meso- (800 m) and bathy- (4400 m) pelagia using primers that are expected to amplify representatives of ~80% of known phylogenetic diversity across all three domains. Comparisons of unique sequences revealed a remarkably low degree of overlap between communities at each depth. The 444,147 sequence tags analyzed represented 62,975 unique sequences. Of these, 3,707 (5.9%) occurred at two depths, and only 298 (0.5%) were observed at all three depths. At this level of phylogenetic resolution, Bacteria diversity decreased with depth but was still equivalent to that reported elsewhere for different soil types. Archaea diversity was highest in the two deeper samples. Eucarya observations and richness estimates are almost one order of magnitude higher than any previous marine microbial Eucarya richness estimates.
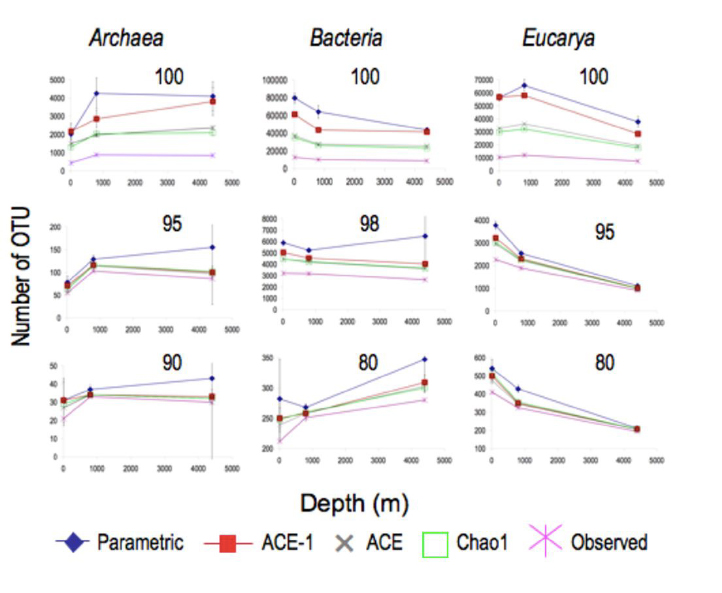
Figure 1. Observed and estimated richness of Archaea, Bacteria and Eucarya, calculated at a variety of phylogenetic levels. Left hand column (a) Archaea, middle column (b) Bacteria, right hand column© Eucarya.
The associations of many Eucarya sequences with putative parasitic organisms may have significant impacts on our understanding of the mechanisms controlling host population density and diversity, and point to a more significant role for microbial Eucarya in carbon flux through the microbial loop. We posit that the majority of sequences detected from the deep sea that have closest matches to sequences from non-pelagic sources are indeed native to the marine environment, and are possibly responsible for key metabolic processes in global biogeochemical cycles.
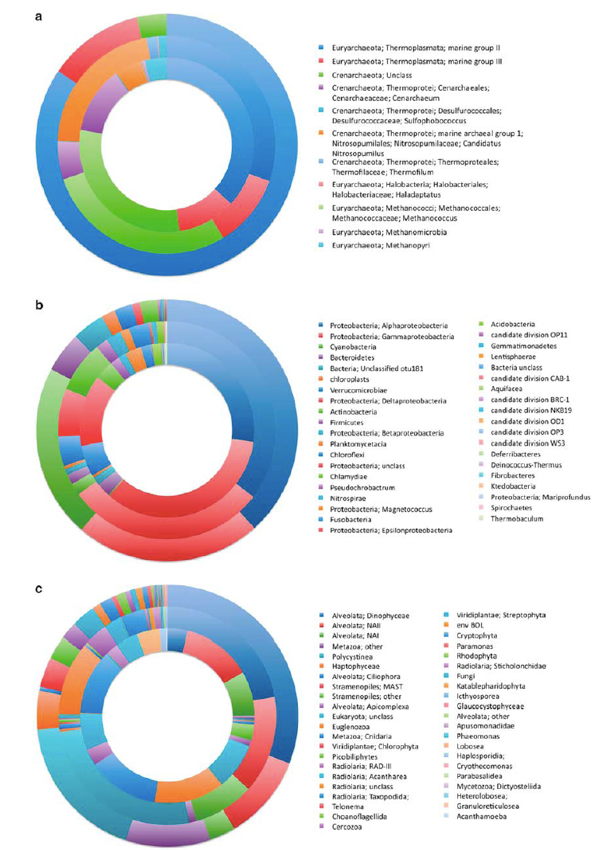
Figure 2. (a) The abundance of major Archaea phylogenetic lineages observed at 10m (outside ring), 800m (middle ring) and 4,400m (inside ring) depths. (b) The abundance of major Bacteria phylogenetic lineages observed at 10m (outside ring), 800m (middle ring) and 4,400m (inside ring) depths. (c) The abundance of major Eucarya phylogenetic lineages observed at 10m (outside ring), 800m (middle ring) and 4,400m (inside ring) depths.
Publications
-
Brown, M. V., Philip, G. K., Bunge, J. A., Smith, M. C., Bissett, A., Lauro, F. M., … Donachie, S. P. (2009). Microbial community structure in the North Pacific ocean. ISME J, 3(12), 1374–1386. doi:10.1038/ismej.2009.86
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Stuart Donachie
Project Investigator
Mark Brown
Co-Investigator
Andrew Bissett
Collaborator
John Bunge
Collaborator
Jed Fuhrman
Collaborator
Federico Lauro
Collaborator
Matthew Smith
Collaborator
Gayle Philip
Postdoc
-
RELATED OBJECTIVES:
Objective 4.1
Earth's early biosphere.