2009 Annual Science Report
 Montana State University
Reporting | JUL 2008 – AUG 2009
Montana State University
Reporting | JUL 2008 – AUG 2009
Computational Chemical Modeling the Link Between Structure and Reactivity of Iron-Sulfur Motifs
Project Summary
Traditionally, the iron-sulfur mineral catalysis, iron-sulfur enzyme catalysis, and biomimetic thrust areas of ABRC have their own unique ways to probe the structure/function relationships at the surface defect sites, at the enzymatic active sites, or at the interface of biomacromolecular and iron-sulfur particle layers, respectively. Computation chemistry can provide a cohesive link among these thrust areas through bridging the enzymatic/mineral catalysis and molecular structure/chemical reactivity via fundamental physico-chemical properties at the molecule level.
Project Progress
A main focus of this thrust was the development a new generation of coordination chemical models for catalytic sites of iron-sulfur minerals, mineral surfaces, and particles. We consider the surface defects of minerals, the perturbed surface structure of iron-sulfur particles embedded in an organic matrix, and the active sites of metalloenzymes as coordination compounds with localized electronic and geometric structures that are being greatly modified by the mineral, organic, and protein environment. This hypothesis allows us to break away from the traditional periodic boundary conditions-based modeling strategies. Using the molecular orbital-based, localized electronic structure description we can employ a large set of theoretical methods that have already been validated for numerous systems. An essential stepping-stone for this work was to utilize virtual chemical models of the FeFe-hydrogenase active site. We have found that protein environment via the orientation of the coordinated amino acid residues can control the cluster’s electron density and thus affecting the cluster electronic and magnetic properties. These protein/cluster interactions also influence the catalytic pathways and rates as key factors in small molecule activation chemistry. A parallel computational work utilizing a multi-edge X-ray absorption spectrochemical library of biomimetic compounds provided the definition of the most reasonable level of theory for molecular size Fe-S motifs.
Using the above insights, we created coordination chemistry-based models for the bulk pyrite and the [100] surface of pyrite. We hypothesize that the bulk environment, similarly to protein environment can be considered as electronic and geometric perturbations to a defect site. By gradually expanding the bulk environment around a central 'active site’ moiety of pyrite, we defined three layers of interactions of the immediately covalently bound 'ligands’ (1st layer), the electrostatic polarizing layer (2nd later), and the terminating bulk with steric constraints (3rd layer) (Figure 1A). By performing a [100] cut to this bulk pyrite model, we can obtain two virtual chemical model of the surface with either S or Fe/S termination as demonstrated in Figure 1B. With the development of these models we are now poised for mapping the potential energy surface of small molecule activation, condensation, and substitution reactions that are relevant to the formation of the building blocks of life.
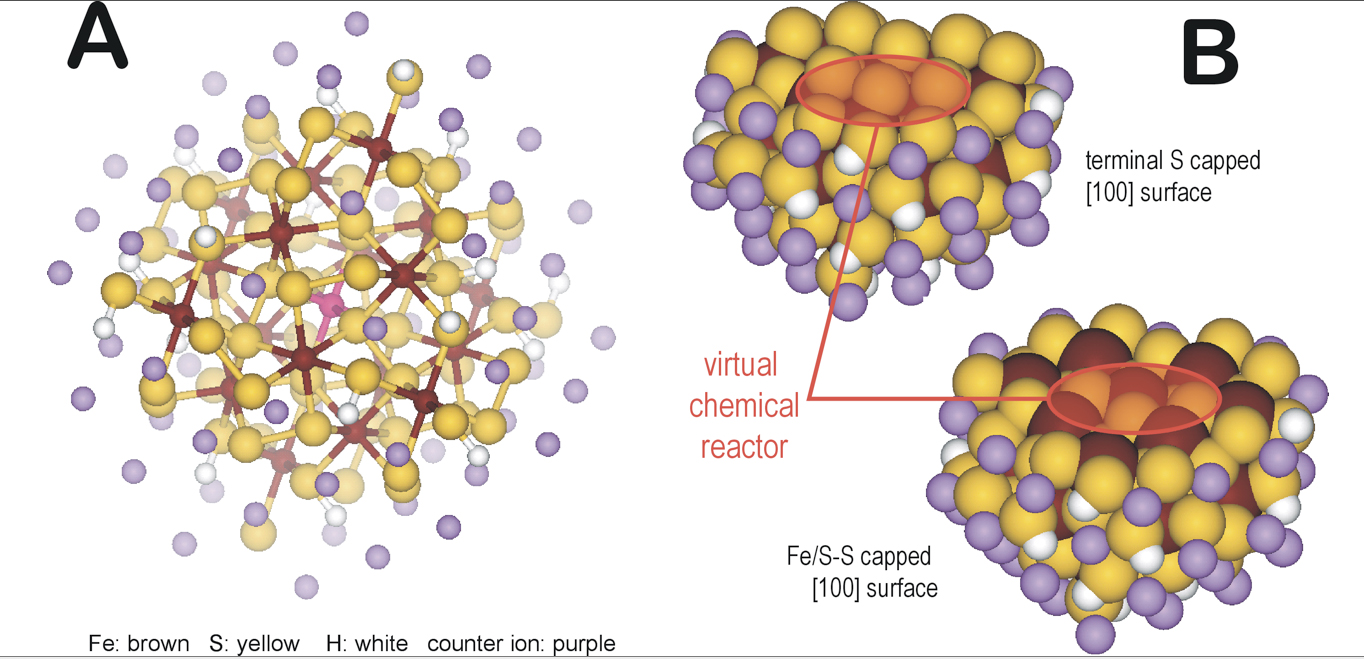
Coordination chemical model-based computational models for pyrite: Non-periodic, coordination chemical model-based computational models for (A) bulk pyrite and (B) pyrite [100] surfaces. The pink atom for (A) represents an Fe(II) center surrounded by six persulfide bonds (1st sphere), that are connected to 18 Fe centers (2nd sphere) and terminated with persulfide and sulfhydryl ligands (3rd sphere) and counter ions (pink atoms) to give a neutral cluster. The surface models (B) were created by an appropriate cut to the bulk model (A) to get [100] surface with sulfur or Fe/persulfide termination. The red circles represent the initial location of a defect site which defined a catalytically active center.
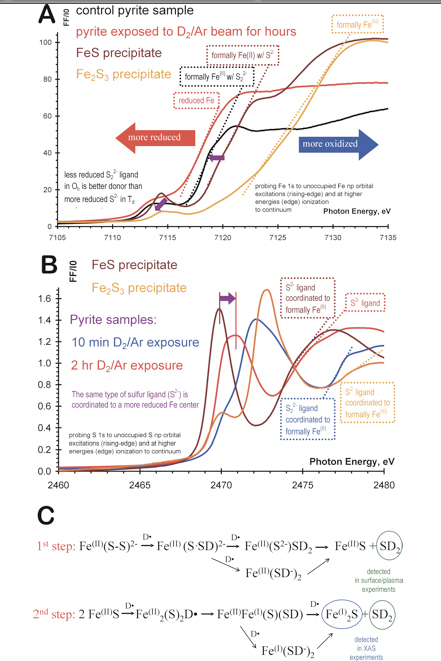
X-ray Absorption Spectroscopy of Fe-S systems: Summary of X-ray absorption spectra at the Fe K-edge (A) and S K-edge (B) and proposed chemical changes© as the result of D2/Ar exposure of pyrite. Both Fe and S K-edge spectra indicate the formation of a reduced Fe-S phase by lower and higher energy rising-edge features, respectively. On the basis of comparison to reference Fe(II) and Fe(III) containing samples, the formal oxidation state can be identified as Fe(I), which is similar to the catalytically active-form of the Fe-S cluster of hydrogenase.
Publications
-
Mulder, D. W., Ortillo, D. O., Gardenghi, D. J., Naumov, A. V., Ruebush, S. S., Szilagyi, R. K., … Peters, J. W. (2009). Activation of HydA ΔEFG Requires a Preformed [4Fe-4S] Cluster. Biochemistry, 48(26), 6240–6248. doi:10.1021/bi9000563
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Alexios Grigoropoulos
Postdoc
Michael Vance
Postdoc
David Gardenghi
Graduate Student
Christopher Koll
Undergraduate Student
Bradley Towey
Undergraduate Student
-
RELATED OBJECTIVES:
Objective 3.1
Sources of prebiotic materials and catalysts
Objective 3.2
Origins and evolution of functional biomolecules
Objective 3.3
Origins of energy transduction
Objective 3.4
Origins of cellularity and protobiological systems
Objective 7.1
Biosignatures to be sought in Solar System materials
Objective 7.2
Biosignatures to be sought in nearby planetary systems
