2015 Annual Science Report
 University of Montana, Missoula
Reporting | JAN 2015 – DEC 2015
University of Montana, Missoula
Reporting | JAN 2015 – DEC 2015
Executive Summary
The question motivating the “Reliving the Past” team is: What forces bring about major transitions in the evolution of complexity? Our research consists of eight projects organized around five questions related to major transitions in the history of life: 1) How do enzymes and metabolic networks evolve? 2) How did the eukaryotic cell come to be, specifically the cell that contained a mitochondrion? 3) How do symbioses arise? 4) How does multicellularity evolve? and 5) How do pleiotropy, epistasis and mutation rate constrain evolution of novel traits? A unifying theme underlying our questions is: how do cooperative vs. competitive interactions play out when independently replicating entities combine to form a larger, more complex whole? Because the principal actors throughout much of life’s history were microorganisms, we approach these questions via experimental microbial genetics, which enables us to enact the evolutionary play repeatedly and from different premises in order to discover why evolution takes certain paths and not others. We seek general principles likely to hold wherever life exists and to collectively affect its degree of complexity.
YEAR 1 RESEARCH HIGHLIGHTS
Theme 1: How do enzymes and metabolic networks evolve?
PROJECT 1: (Shelley Copley, Colorado-Boulder) When microbes face an environmental challenge or opportunity, pre-existing enzymes with promiscuous secondary activities can be recruited to provide newly important catalytic functions. Mutations that increase the efficiency of a new activity often compromise the original activity, resulting in an inefficient bifunctional enzyme. Under such conditions, duplication/amplification of the gene encoding the “weak-link” enzyme can increase its expression and consequently increase fitness. Mutations that occur in various copies of the gene can improve the level of one or the other of the essential activities. As improved enzymes evolve, the number of gene copies will decrease, leading ultimately to a pair of genes encoding specialized enzymes. The existence of large superfamilies of enzymes attests to the importance of this process. However, the dynamics of the process itself are poorly understood. The Copley team at Colorado is examining the process of gene duplication/amplification and divergence in the case of E. coli E383A ProA, which can serve the functions of both ProA (essential for proline biosynthesis) and ArgC (essential for arginine biosynthesis), albeit poorly (see Figure 1). Since both activities are essential for growth on glucose, E383A ProA is the ‘weak-link” enzyme that limits growth rate in a strain that lacks argC. Shelley’s group has previously shown that growth rate can be increased by massive gene amplification, by point mutations in the promoter, and by a synonymous mutation in the upstream gene in the operon that creates a new transcriptional start site for the gene encoding E383A ProA. Using an experimental evolution approach Copley’s team will follow the appearance of mutations in various alleles of an amplified array and characterize the encoded enzymes throughout the process of divergence to two specialized enzymes.

PROJECT 3: (Scott Miller, Montana) The Miller group is also exploring the evolutionary dynamics of duplicated genes, albeit those closely involved with exchange of genetic information via homologous recombination (HR). In bacteria HR is mediated by the generalist enzyme, RecA. The cyanobacterium Acaryochloris is unusual in having multiple copies of recA, which is essential for maintenance of genome stability, but is also involved in gene duplication. Significantly, Acaryochloris genomes are among the largest observed for bacteria (>8 Mbp), and are thought to be the product of genome expansion arising from high rates of gene duplication. Bacterial genomes typically harbor few duplicates, however, using the amount of synonymous divergence between duplicates at degenerate codon positions (dS) as a proxy for age, Miller has shown that Acaryochloris genomes are specifically enriched for recent duplicates. In Year-1 Scott’s team developed tools to study Acaryochloris RecAs in the genetic model bacterium E. coli, which they have used to demonstrate functional divergence among duplicated RecAs. The Miller Lab is collaborating with Michael Cox (University of Wisconsin) to functionally dissect divergent Acaryochloris RecA proteins in their purified forms.
Theme 2: How did the eukaryotic cell come to be, specifically the cell that contained a mitochondrion? and Theme 3: How do symbioses arise?
Project 2: (John McCutcheon, Montana) The origins of mitochondria and chloroplasts are two of the great, unsolved mysteries in biology. While it is clear that these organelles used to be bacteria, the evolutionary paths they took as they transitioned from bacteria to organelle are poorly understood as they occurred >1.5 billion years ago. Similar to a matryoshka doll, insects like cicadas host endosymbionts that themselves have symbioses with other bacteria. Because these more recent symbioses resemble organelles, John McCutcheon’s group at Montana studies them to better understand organelle evolution. A milestone in this investigation was McCutcheon’s landmark paper, “Genome expansion via lineage splitting and genome reduction in the cicada endosymbiont Hodgkinia” in PNAS (doi: 10.1073/pnas.1421386112; Cover of 08/18/15). What this paper describes is a process whereby the original single bacterial endosymbiont lineage has split into several related lineages inside the cicada (Figure 2). Each individual new genome has become smaller through the expected process of endosymbiotic genome reduction. However, critically, from the perspective of the host insect, the collective endosymbiotic genome has actually grown in size because the number of lineage splitting events has outpaced the genome reductive process found in individual lineages. Thus, genome expansion through lineage splitting and genome reduction.

This is interesting on its own, but has a fascinating parallel to mitochondrial genome evolution (Figure 3). In some eukaryotic lineages, in particular plants and fungi, mitochondrial genomes have undergone similar fragmentation and genome expansion events. While the mechanism of expansion seems different in each case (fungi, plants, and cicada endosymbionts), we hypothesize that they are united in being a result—a syndrome, perhaps—of long-term obligate host association. It has been argued in mitochondrial cases that this is an example of non-adaptive genome expansion, and we favor that idea in our case, too. Collectively these systems are remarkable test cases to study complexity in biology that arises not because it makes the organism more fit, but simply because the organism cannot stop it from happening.

PROJECT 4: (Margie Kinnersley, Montana) NPP Fellow Kinnersely is using an experimental evolution approach to study organelle evolution. Models of mitochondriogenesis agree that the proto-mitochondrion was an α-proteobacterium that became associated with another cell. However, models differ with respect to the host’s phylogenetic affiliation and the manner in which the mitochondrion was acquired. “Mitochondrion-late” models posit that the host was an amitochondriate eukaryote that engulfed a proto-endosymbiont via phagocytosis. “Mitochondrion-early” models propose that the fusion event occurred between a prokaryotic host and an α-proteobacterium. Opponents of mitochondrion-early models argue that the host could not have been a prokaryote because prokaryotes are incapable of phagocytosis. Using the bacterial parasite Bdellovibrio bacteriovorus and its host Escherichia coli Kinnersley is testing Margulis’ hypothesis that the proto-mitochondrion was a predator that actively entered the host cell rather than being passively ingested. Her experiments select for two essential characteristics of mutualistic endosymbioses: (i) commitment of the parasite to existence inside its host, and (ii) positive reinforcement of long-term coexistence via benefits to both partners. In Year-1 she has succeeded in tagging both species with different colored Fluorescent Proteins, which will enable Bb-Ec endosymbionts to be selected by FACS during experimental evolution, and she has genetically engineered host-parasite interdependencies based on complimentary amino acid biosyntheses (much like what naturally evolved in McCutcheon’s Hodgkinia spp.) and antibiotic resistances.
Theme 4: How does multicellularity evolve?
PROJECT 5: (Vaughn Cooper, University of Pittsburgh) Biofilms are surface-associated communities that represent the dominant form of microbial life and likely reflect the origins of multicellularity. Surface growth can preserve diversity by allowing multiple lineages to evolve independently. When these subpopulations expand and reestablish contact, their interactions can range from antagonistic to synergistic, in the latter potentially facilitating mutualistic symbiosis (related to Theme 3). For example, biofilm overproducers can engineer their environment and facilitate the growth of and attachment of others. Alternatively, subpopulations that adapted by similar mechanisms could compete to their mutual detriment. The forces that govern the balance of these interactions are mostly unknown yet are central to understanding the evolution of biocomplexity. Vaughn Cooper’s group, which relocated in Year-1 to the Univ of Pittsburgh School of Medicine, experimentally studies how biofilm communities evolve in opportunistic pathogens (see Figure 4).

In Year-1 Cooper’s team reported how long-term experimental evolution of Burkholderia cenocepacia biofilms consistently results in character displacement, enabling different ecotypes to occupy different niches (Ellis et al. 2015. Evolution 69:283-93). Character displacement (Figure 5) was evident early in each of the replicate experiments. Remarkably, over evolutionary time, niche partitioning resulted in synergistic communities that were both more competitive and more productive than the clonal ancestor from which they arose. The mechanistic basis of these synergistic interactions appears to be related to earlier colonization by the minority rough® and wrinkly (W) types as well as by their production of metabolic byproducts that together help support the dominant studded (S) type, providing a soon-to-be classic example of the experimental evolution of facilitation.

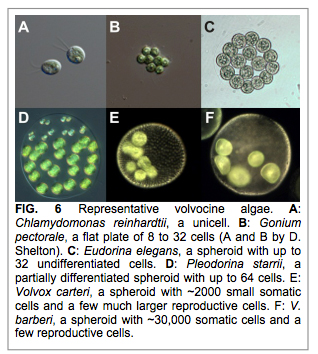
PROJECT 6: (Matt Heron, Montana) Multicellularity has evolved independently in at least 25 separate lineages, including the Eubacteria, Archaea, and multiple lineages spanning the deepest divergences within the eukaryotes. Independent origins of multicellularity are replicate experiments with the potential to reveal general principles involved in this transition. Unfortunately, for most lineages the evidence of the intermediate stages has been decimated by extinction and obscured by the limitations of the fossil record. However, in one lineage – the volvocine green algae (Fig. 6) – abundant evidence remains in the form of extant species with various mixtures of derived and ancestral traits.
Former NPP Fellow and now Research Assistant Professor Matt Herron is carrying out experimental evolution on the unicellular alga Chlamydomonas reinhardtii, imposing selection for unicells to form – and heritably maintain – larger sized clusters. One form of selection, centrifugation, is simple and arbitrary, favored an alternating uni- and multicellular life cycle (Ratcliff, Herron et al. 2013 Nature Communications 4, 2742-2742). To gain insight into the underlying genetic basis of this novel trait Herron has followed up on these experiments with an RNA-Seq analysis of the life cycle of ancestral and derived (multicellular) C. reinhardtii. His team has also performed bulked segregant analysis of multicellular F2 progeny from this experiment, which has revealed two chromosomal regions that are substantially enriched for alleles from the multicellular strain in the multicellular pool (Figure 7). Among these are several genes involved in the production of extracellular matrix and one known to function in cell cycle control.

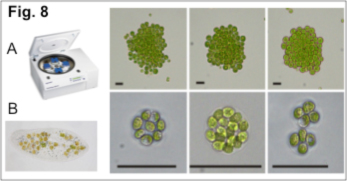
Herron’s team is also analyzing evolution experiments where large size was selected for by culturing Chlamydomonas with Paramecium. Whereas centrifugation experiments selected for amorphous multicellular clusters (Figure 8A below), predation pressure resulted in cell wall enclosed multicellular C. reinhardtii (Figure 8B) that are reminiscent of extant Volvocine algal taxa such as Gonium and Eudorina (Figure 6B, C).
Theme 5: How do pleiotropy, epistasis and mutation rate constrain the evolution of novel traits? A population’s tempo and trajectory of evolutionary change are not only subject to ecological forces like predation and competition but also to intrinsic factors such as rates of mutation and recombination as well as constraints imposed on novel mutations by how well they interact with other genes. For example, no matter how advantageous a novel trait may be under particular circumstances, it may be difficult to evolve that trait if the mutant gene producing that trait “plays poorly” with other genes, reducing overall fitness. In order to succeed in our quest to “relive the past” in laboratory we are taking stock of such factors in two model systems: the bacterium Escherichia coli (Project 7) and the eukaryote Saccharomyces cerevisiae (Project 8).
PROJECT 7: (Phil Gerrish, currently at Montana, Paul Sniegowski, UPenn) The UPenn team led by Sniegowski and Gerrish are examining the evolutionary relationship between rates of genetic mutation and genetic recombination. Their work, which has both theoretical and experimental components, raises general questions about the stability of heredity and the implications of that stability for adaptive process. In Year-1 they have made progress on multiple fronts, of which we offer two examples. They have explored in silico the effects of deleterious mutations on the fate of beneficial mutations in evolving asexual populations; these studies show that the likelihood that even a strongly beneficial mutation goes to fixation is strongly diminished if error rate exceeds a certain threshold (Figure 9).

While DNA polymerases of all species have intrinsic error rates that are relatively high (e.g., 10-6 bp per generation), populations’ effective mutation rates are usually low (e.g., 10-10 bp per generation), owing to a variety of DNA repair enzymes that correct errors that escape proofreading. A defect arising in any one of the genes that encode such enzymes can result in a “mutator,” a lineage whose mutation rate is orders of magnitude higher than its wild-type progenitor. In an experimental study UPenn team and their NAI collaborator Ben Galeota-Sprung have investigated the evolutionary fate of mutators in populations of E. coli genetically engineered to have high- and low recombination rates. They discovered that mutator alleles quickly fix in low-recombination strains via genetic hitchhiking, but that high-recombination impedes this process (Figure 10).

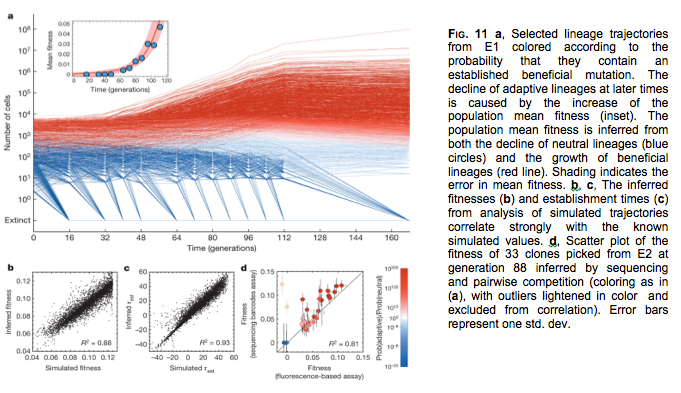
PROJECT 8: (Gavin Sherlock, Stanford) Even as the UPenn team is studying the interplay between mutation rate, recombination rate and the likelihood with which novel alleles become fixed, the Stanford team aims to shed light on the rates at which deleterious, neutral and beneficial mutations accumulate, and the spectrum of their fitness effects in different genetic backgrounds and environmental contexts. Detailed understanding of these parameters is required to ascertain a novel trait’s “evolvability.” For example, in one genetic background a novel allele, and its associated trait, may be beneficial, but in another it may be detrimental owing to pleiotropy or epistatic interactions with other genes. Likewise, the fitness effect of a given mutation may critically dep end on the environment, such that a mutation that is beneficial in one setting but detrimental in another. In Year-1 the Stanford team achieved a major breakthrough. Their study, “Quantitative evolutionary dynamics using high-resolution lineage tracking” published in Nature (doi:10.1038/nature14279) describes a sequencing-based ultra high-resolution lineage tracking system in Bakers yeast that enabled then to monitor, simultaneously and in situ, the relative frequencies of ~500,000 DNA barcoded lineages as they evolved in the lab (Figure 11). This technological feat, based on a plan of attack outlined in our CAN-7 proposal, represents the first time the beneficial mutation rate and the distribution of fitness effects (DFE) has been measured empirically, in real time in an evolving population of any species. This breakthrough will enable co-I Sherlock and his team to make rapid progress in Year-2 towards achieving their NAI research aims.
Theoretical Integration (Eric Smith, ELSI and Montana). As evolutionary transitions are defined by increases in interdependence and autonomy these five research themes are being integrated by modeling interactions as new levels of selection form. In Year-1 co-I Smith has focused on aspects of the role of ecosystem feedback in determining the relations among fitness functions and the co-evolutionary dynamics of multiple genomes. Specifically, he is developing modeling frameworks in which to address two questions related to theme of cooperating genomes under selection, as being explored in different ways by McCutcheon, Kinnersley, Cooper and and Herron in Projects 2, 4, 5 and 6, respectively. When are two heads (i.e., genomes) better than one? and, What are the co-evolutionary dynamics on stoichiometrically-constrained spaces? The first of these questions is organismal, the second chemical. The second models ecological feedbacks, derived from the reaction-network theory of alternative implementations of the pentose-phosphate pathway. In actual ecosystems, coevolution is constrained by the reliance of organisms on metabolites (or their avoidance of toxic effects from wastes) that only they or other organisms in the ecosystem can produce or recycle. The fitnesses of all member species depend on the frequencies of all others, not directly as through predation, but mediated by environmental concentrations of metabolites that have their own dynamics. The coupled rates of production of metabolites and wastes within each organism lead to complex webs of constraint; and the determination of balanced flows – the only class of solutions that are eligible to be evolutionary steady states – is a computationally challenging.
Co-I Smith’s work is highly interdisciplinary and enlarges the scope of our CAN-7 NAI enterprise to include investigators at ELSI and elsewhere. For example, the use of stoichiometric models of ecological feedbacks on fitness has benefitted expressly from a collaboration with computer scientists and computational chemists based at the University of Vienna (Christoph Flamm), the University of Leibzig (Peter Stadler), the University of Southern Denmark (Daniel Merkle), and most recently hired at ELSI in Tokyo (Jakob Lykke Andersen). These researchers are the innovators of sophisticated graph-grammar methods (based on category theory) for the automatic generation of reaction networks of arbitrary size from finite suites of generating mechanisms. Their methods to not only generate networks of arbitrary complexity, but also solve for the sets of all balanced flows on these networks, enables exact formal modeling of the kinds of complex stoichiometric constraints that exist both within metabolisms, and among organisms exchanging essential metabolites within ecosystems, which would not be possible at present from empirical data due to limitations on the completeness of measurements and meta-metabolism models at the ecosystem level for syntrophic communities.
One of co-I Smith’s major Year-1 achievements was publication of the monograph: “Symmetry and Collective Fluctuations in Evolutionary Games” by E. Smith & S. Krishnamurthy (2015). Institute of Physics Press, Bristol, 303 p. doi:10.1088/978-0-7503-1137-3
Related research activities
Our team is building on the foundation of the NAI cooperative agreement. Collectively in Year-1 we submitted 30 new grant proposals. While most of these are still pending, 7 new awards have been made totaling almost $10M. Montana CAN7 researchers and their students have given 65 invited seminars, including 4 presentations at the Gordon Research Conference on Microbial Population Biology, and 8 talks at the Chicago AbSciCon meeting June 2015.
Publications
- Herron, M. D. (In press) Cells, colonies, and clones: individuality in the volvocine algae, in: E pluribus unum: bringing biological parts and wholes into historical and philosophical perspective, L. Nyhart and S. Lidgard, eds., University of Chicago Press, Chicago.
- Smith, E. and S. Krishnamurthy (2015) Symmetry and Collective Fluctuations in Evolutionary Games. Institute of Physics Press, Bristol, 303 p.
-
Bennett, G. M., McCutcheon, J. P., McDonald, B. R., & Moran, N. A. (2015). Lineage-Specific Patterns of Genome Deterioration in Obligate Symbionts of Sharpshooter Leafhoppers. Genome Biology and Evolution, 8(1), 296–301. doi:10.1093/gbe/evv159
-
Calderón-NoreñA, D. M., González-Novo, A., Orellana-Muñoz, S., Gutiérrez-Escribano, P., Arnáiz-Pita, Y., Dueñas-Santero, E., … Vázquez De Aldana, C. R. (2015). A Single Nucleotide Polymorphism Uncovers a Novel Function for the Transcription Factor Ace2 during Candida albicans Hyphal Development. PLOS Genetics, 11(4), e1005152. doi:10.1371/journal.pgen.1005152
-
Campbell, M. A., Van Leuven, J. T., Meister, R. C., Carey, K. M., Simon, C., & McCutcheon, J. P. (2015). Genome expansion via lineage splitting and genome reduction in the cicada endosymbiont Hodgkinia. Proc Natl Acad Sci USA, 112(33), 10192–10199. doi:10.1073/pnas.1421386112
-
Cooper, V. S., Staples, R. K., Traverse, C. C., & Ellis, C. N. (2014). Parallel evolution of small colony variants in Burkholderia cenocepacia biofilms. Genomics, 104(6), 447–452. doi:10.1016/j.ygeno.2014.09.007
-
Ellis, C. N., Traverse, C. C., Mayo-Smith, L., Buskirk, S. W., & Cooper, V. S. (2015). Character displacement and the evolution of niche complementarity in a model biofilm community. Evolution, 69(2), 283–293. doi:10.1111/evo.12581
-
Hallam, S. J., & McCutcheon, J. P. (2015). Microbes don’t play solitaire: how cooperation trumps isolation in the microbial world. Environmental Microbiology Reports, 7(1), 26–28. doi:10.1111/1758-2229.12248
-
Herron, M. D. (2016). Fitness and Individuality in Complex Life Cycles. Philosophy of Science, 83(5), 828–834. doi:10.1086/687867
-
Herron, M. D. (2016). Origins of multicellular complexity: Volvox and the volvocine algae. Mol Ecol, 25(6), 1213–1223. doi:10.1111/mec.13551
-
Herron, M. D., & Nedelcu, A. M. (2015). Volvocine Algae: From Simple to Complex Multicellularity. Advances in Marine Genomics, None, 129–152. doi:10.1007/978-94-017-9642-2_7
-
Keeling, P. J., McCutcheon, J. P., & Ford Doolittle, W. (2015). Symbiosis becoming permanent: Survival of the luckiest. Proc Natl Acad Sci USA, 112(33), 10101–10103. doi:10.1073/pnas.1513346112
-
Levy, S. F., Blundell, J. R., Venkataram, S., Petrov, D. A., Fisher, D. S., & Sherlock, G. (2015). Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature, 519(7542), 181–186. doi:10.1038/nature14279
-
Nozaki, H., Ueki, N., Misumi, O., Yamamoto, K., Yamashita, S., Herron, M. D., & Rosenzweig, F. (2015). Morphology and reproduction of Volvox capensis (Volvocales, Chlorophyceae) from Montana, USA. Phycologia, 54(3), 316–320. doi:10.2216/15-14.1
-
O’Rourke, D., FitzGerald, C. E., Traverse, C. C., & Cooper, V. S. (2015). There and back again: consequences of biofilm specialization under selection for dispersal. Frontiers in Genetics, 6. doi:10.3389/fgene.2015.00018
-
Scharf, C., Virgo, N., Cleaves, H. J., Aono, M., Aubert-Kato, N., Aydinoglu, A., … Yabuta, H. (2015). A Strategy for Origins of Life Research. Astrobiology, 15(12), 1031–1042. doi:10.1089/ast.2015.1113
-
Schep, A. N., Buenrostro, J. D., Denny, S. K., Schwartz, K., Sherlock, G., & Greenleaf, W. J. (2015). Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res., 25(11), 1757–1770. doi:10.1101/gr.192294.115