2015 Annual Science Report
 University of Illinois at Urbana-Champaign
Reporting | JAN 2015 – DEC 2015
University of Illinois at Urbana-Champaign
Reporting | JAN 2015 – DEC 2015
Project 5: Control of Evolvability and Chromosomal Rearrangement by Stress
Project Summary
Using a bacterial system, we are investigating the regulation of genomic change by stress, both small changes in DNA sequence and larger chromosomal rearrangements. That mutation rates should be regulated by stress has wide significance for understanding mechanisms of evolution. Our major goal in this project is to discover how the general stress-response regulator switches from accurate modes of DNA repair to error-prone modes. We have established that mutation under stress only occurs if there are replicating blocking lesions in DNA, usually formed by reactive oxygen. We have shown apparent positive and negative regulation of the levels of oxygen radicals. We are now working to discover the origin of the radicals so that we can ask whether radicals form as an unavoidable byproduct of respiration, or whether this represents a dedicated mechanism to instigate mutation, and hence evolution.
Project Progress
During 2014, we had shown that nucleoid associated proteins regulate both single nucleotide change and chromosomal rearrangement under stress in trans rather than by regulating access of DNA repair proteins to the DNA, where they might cause genetic change. This work is now published.
In the course of that study, we found that both single nucleotide variants (SNV) and gross chromosomal rearrangements (GCR) depend on oxidative damage to the cell, having found both positive and negative regulation of the availability of reactive oxygen species (ROS). We followed up on this unexpected and important discovery by showing that ROS-induced lesions in DNA are required for both SNV and GCR, that the oxidative damage needs to be in DNA, not protein or RNA, that the effects of ROS damage are not mediated through mismatch repair saturation or double-strand break formation, that SNVs do not occur if the lesions are excised by base excision repair (BER) and that we could substitute other replication-blocking base damage for oxidative damage.
During 2015, we have refined these data, performed additional controls and extended our understanding in several ways. First, we find that the oxidized nucleoside 8-oxo-dG is incorporated into DNA from the nucleotide triphosphate pool, not formed by direct oxidation of DNA. Thus, we show that mutation rate is regulated by the composition of the nucleotide pool. We also show that over-expression of the error-prone DNA polymerase DinB, is not sufficient to cause mutation. Base damage is also required.
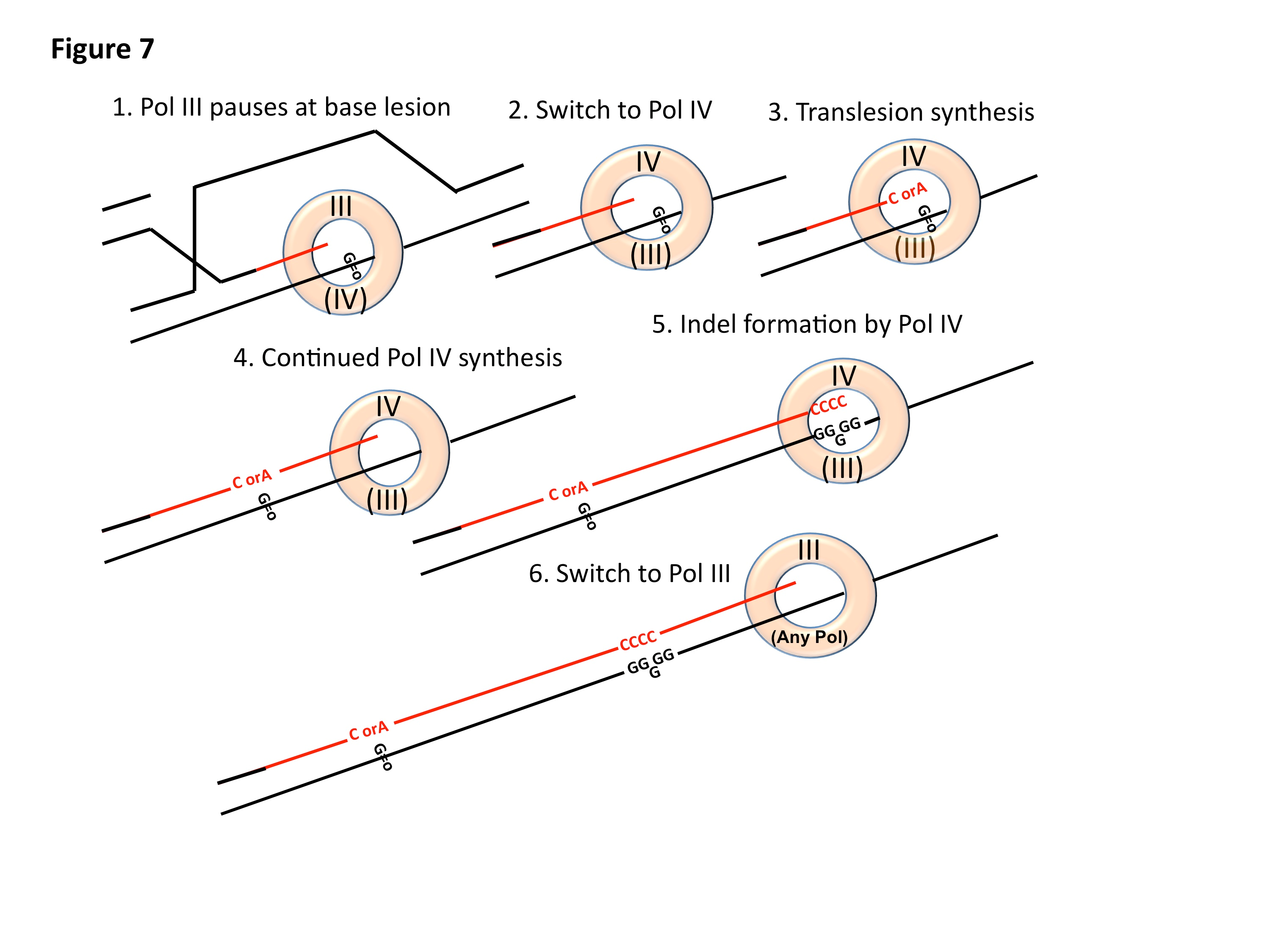
We have developed a model to explain the effects of ROS on indel mutation in stressed cells. It had been thought that over-expression of error-prone polymerases was mutagenic because of polymerase competition. We now offer the idea that the replicative polymerase Pol III is so processive that polymerase switching will be very rare, unless something happens to cause the replicative polymerase to pause. There are two polymerase-binding sites on the -clamp in the replisome, one of which holds the active site polymerase. Upon pausing, the polymerase on the inactive binding-site moves to the active site and takes over replication. If this second polymerase is DinB, replication will now be able to cross and/or extend from the base lesion. This is mutagenic in two potential ways. One is that because DinB has a large template-binding site, it can accept 8-oxo-dG in an alternative isomeric form that will pair with adenine instead of cytosine. Thus 8-oxo-dG is replicated to form a GA base pair in about 20% of replications, causing a G to T transversion mutation. Second, because DinB is mildly processive, it will still be in use for some distance after it has crossed the lesion. Its large active site allows the formation of extrahelical bases, which, when replicated give deletions of one base pair. These indels are the readout for stress-induced mutagenic break repair that we use in these assays. Because we also require ROS to form GCR in this assay, we postulate that Pol I (required for GCR in this assay) will be the polymerase to take the active site where it can give GCR by template switching, a know activity of A-family polymerases. This work is now ready for publication.
The requirement for ROS damage to remain in DNA for indel formation applies to both growing and starved cells. Thus, this is not a specific property of adaptive mutation under stress. We have fairly good evidence that substitution mutation increases if we give more glycosylase activity by over-expression of MutM, so the explanation of the mechanism of indel formation given above does not apply. Substitution mutation is enhanced by increased BER. We have found, then, that BER is a mutagenic repair mechanism, and this mechanism is responsible for a large portion of spontaneous substitution mutation both in growth and under stress, We do not yet know whether this comes from gap-filling after excision of a damaged base followed by AP endonuclease action, or from translesion synthesis across an apurinic site in DNA after excision. We also want to know whether our findings for stationary phase starved stressed cells also applies to non-stressed growing cells. To achieve this we over-express MutM, the glycosylase that excises 8-oxoG from DNA in the course of BER. We have developed new assays to determine these parameters, and we do not yet have full data. These findings apply to SNV formation, not to GCR, for which we have now developed a new assay. We plan to complete the study of the causes of spontaneous mutation, and then proceed to a similar study of the basis of chromosomal rearrangement.
Given that we have shown that there is regulation, both positive and negative, of the availability of ROS, we will also ask whether mutation is being directed to stressed cells by increases in the availability of ROS under starvation. Specifically, we are asking whether stress responses change cellular physiology to generate ROS, a possible mechanism for enhancing mutability at times when it would be advantageous to allow cells to adapt to the stress-provoking environment. If we find that the general stress response regulates ROS level, we might have answered the question as to how stress regulates mutation.
Publications
-
(no authors found) (2016). Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria. []. doi:10.1002/9781119004813
-
Moore, J. M., Magnan, D., Mojica, A. K., Nunez, M. A. B., Bates, D., Rosenberg, S. M., & Hastings, P. J. (2015). Roles of Nucleoid-Associated Proteins in Stress-Induced Mutagenic Break Repair in Starving Escherichia coli. Genetics, 201(4), 1349–1362. doi:10.1534/genetics.115.178970
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Susan Rosenberg
Co-Investigator
Jessica Moore
Collaborator
Philip Thornton
Collaborator
Raul Correa
Postdoctoral Fellow
-
RELATED OBJECTIVES:
Objective 5.1
Environment-dependent, molecular evolution in microorganisms
