2013 Annual Science Report
 University of Wisconsin
Reporting | SEP 2012 – AUG 2013
University of Wisconsin
Reporting | SEP 2012 – AUG 2013
Project 2C: Calibrating the [^13^]C-[^18^]O (“clumped”) Isotope Temperature Scale
Project Summary
Determining paleotemperatures in ancient fluid-mineral systems is key to determining ancient habitability. Stable oxygen isotope studies of carbonates have long used changes in 18O/16O ratios to infer the temperature from which carbonate precipitated, using a laboratory-calibrated temperature conversion, but this requires knowledge of the 18O/16O ratios of the fluid. This is often not known. A relatively new approach is to use the non-random variations in rare C and O isotopes, specifically the preferential enhancement of 13C-18O bonds, which has been shown to be related to temperature and independent of the fluid isotopic composition. Experimental calibrations, however, have been inconsistent, and goal of this project is to reconcile these discrepancies.
Project Progress
Stable isotope compositions generally consider substitution of a single minor isotope, such as 13C16O16O or 12C18O16O in carbon dioxide because multiply-substituted minor isotopes such as 13C18O16O are very low in abundance in nature. Recently, however, the unique aspects of multiply-substituted minor isotopes have been exploited (Eiler, 2011). Now termed “clumped-isotope” geochemistry, to describe, for example, the enhanced stability of 13C-18O bonds in CO2 and carbonates relative to a random or stochastic distribution of minor isotopes, this field of stable isotope geochemistry promises great breakthroughs in paleoclimate studies that require accurate determination of paleotemperatures, as well as the study of atmospheric gases. For CO2 and carbonate, the measured enrichment in 13C-18O bonds relative to that expected for a random distribution of 13C-18O bonds is defined as Δ47, reflecting nominal mass 47 for 13C18O16O, and this has been shown to be an inverse function of temperature. Recent work on biogenic calcite and aragonite utilizing clumped isotopes continues to reveal fractionation and clumping relations that do not seem to be consistent with many inorganically precipitated carbonates or theoretical calculations (Eagle et al., 2013; Saenger et al., 2012; Thiagarajan et al., 2011; Tripati et al., 2010), indicating a new experimental approach may be fruitful.
In addition, expanding our understanding of natural and biogenic isotopic dynamics and fractionations occurring in the H2O-CO2-carbonate system is also highly desirable considering recent discoveries related to water, CO2, and carbonate formation on Mars (Halevy et al., 2011) and suspected connectivity between early Earth climate and microbial evolution (Pierrehumbert et al., 2011 and references therein).
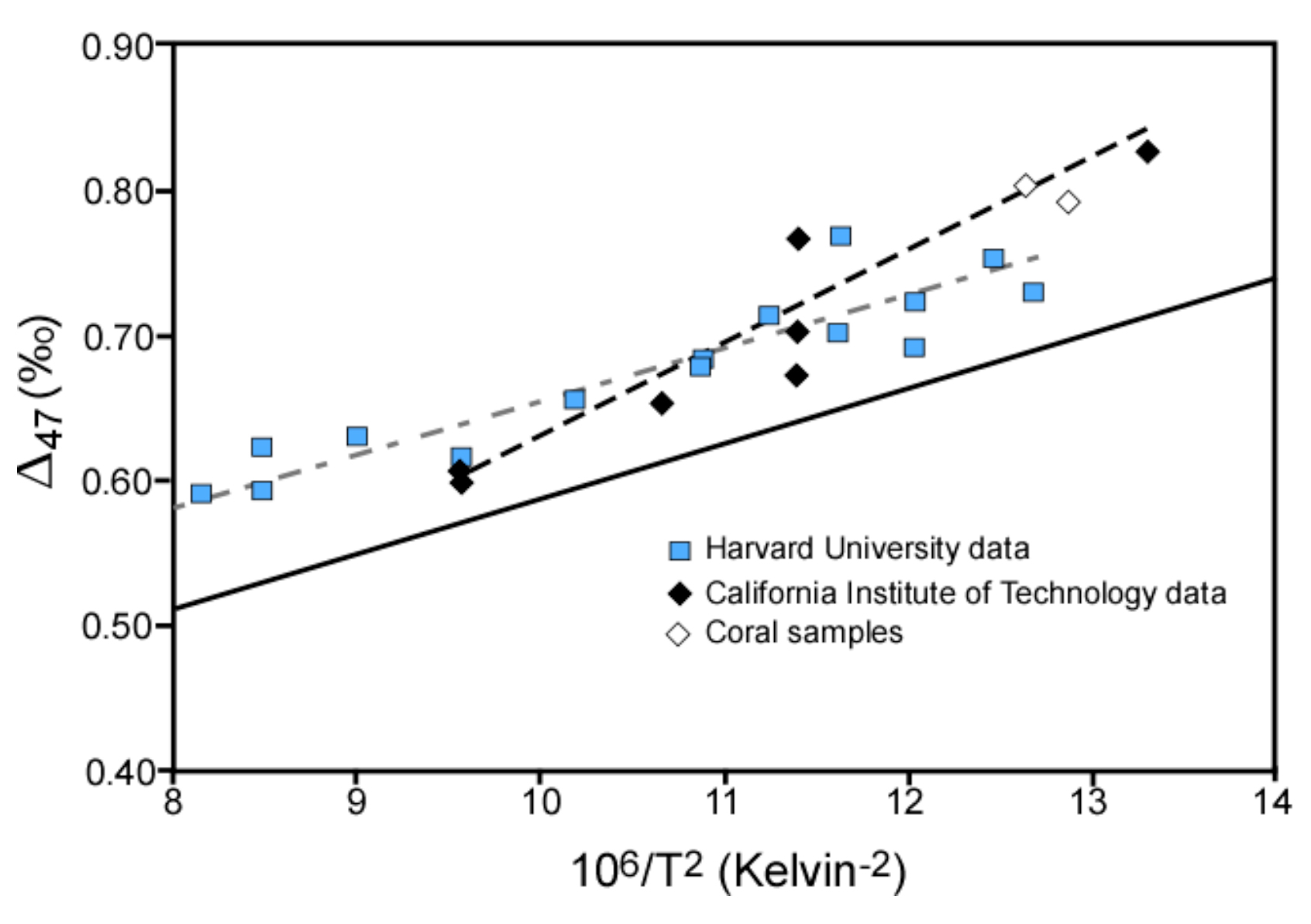
Figure 1 illustrates temperature-clumped isotope relations for two synthetic calibrations, a theoretical calculation, and biogenic carbonate material (coral). Theories used to explain variable isotope patterns observed in different types of carbonate material focus on precipitation conditions including CO2-H2O chemistry, system pH, and formation rate. This project explores probable causes of discrepancies among clumped isotope signals found in biogenic, inorganic, and synthetic carbonates and aims to decouple simultaneous physiochemical, thermodynamic, and kinetic processes that can all act on systematic partitioning of isotopes during carbonate formation.
We have adopted a CO2-H2O equilibration system to use in conjunction with carbonate precipitation experiments. The paring is designed to dynamically mix a constant stream of gas-phase CO2 with liquid phase H2O in order to fully exchange oxygen atoms and bring CO2 into isotope as well as thermal equilibrium with the water used to form solid carbonate minerals. The equilibration system will circumvent CO2-H2O reactions suspected to impart non-equilibrium isotope and thermodynamic offsets observed in kinetically controlled inorganic precipitations as well as some types of biogenic vital effects.
In Year 1, we evaluated the efficiency of the new CO2-H2O equilibration system as a function of time and under variable system conditions (Figure 2). The oxygen isotope composition of system water as well as system CO2 are both plotted. Additionally, the theoretically calculated δ18O value of CO2 in equilibrium with the water is also included. Temperature is plotted against the right-hand ordinate axis in Figure 2a and percent oxygen exchange is plotted on the right-hand ordinate axis in Figure 2b. Not indicated in the figure are changes in partial pressure of CO2 from 0.2 to 15 %, which were also investigated.

The primary variable influencing establishment of oxygen isotope equilibrium in this system is the magnitude of change in δ18O for CO2 traveling through the equilibration system. This composition change is a function of variables such as temperature, heterogeneous phase exchange, and the isotope composition of system water. The ratio of water 18O/16O in the system fluctuates slightly on a day-to-day basis due to loss from evaporation and addition of makeup water. However, a major change in the oxygen isotope composition of the system water (about 10 ‰) was induced for approximately the last quarter of evaluation.
The influence of changing system water can be observed in Figure 2 by the simultaneous shift in CO2 and H2O to higher δ18O values on 8/7/13. All experiments conducted with system water of δ18O values in the 6 to 7.5 ‰ range failed to reach equilibrium despite CO2 changes in δ18O of up to 13 ‰ over the course of the experiment. CO2 oxygen isotope exchange dipped down to 63 % after the initial water change but rebounded to about 90 % before the system temperature was increased.
Most experiments conducted with system water in the δ18O = -6.5 to -5 ‰ range and above 13°C reached or at least closely approached equilibrium. That is, the measured oxygen isotope compositions were within ~10 % of the calculated value. CO2 processed at temperatures near 5°C, however, were found to be lower by up to 0.8 ‰. Taking the starting isotope composition of CO2 into account, these gasses only reached 30 % of their full equilibration value. Isotope ratio offsets at reactions occurring at low temperatures indicate that reaction temperature is an important secondary factor in determining the time necessary to reach isotope equilibrium between CO2 and H2O in this system.
The oxygen isotope exchange experiments described here were conducted with a single equilibration “unit” with a residence time of approximately 2.5 hours. However, multiple units were tested for functionality. Individual units can be connected in series in order to double, triple, etc. the residence time of CO2 in the equilibration system. While oxygen isotope equilibrium was not reached between gas-phase CO2 and liquid water under many of the experimental conditions described here, it should be the case that prolonging residence time by connecting two or more units in series will provide the reaction time necessary to reach isotope equilibrium. This approach has been adopted in order to prevent kinetic fractionation between CO2 and H2O during chemostat carbonate synthesis experiments. In Year 2 we will use this new apparatus to perform clumped-isotope experiments that will use seed crystals, an approach that is designed to minimize kinetic isotope effects.
References Cited
Dennis KJ & Schrag DP (2010) Clumped isotope thermometry of carbonatites as an indicator of diagenetic alteration. Geochim. Cosmochim. Acta 74(14):4110-4122.
Dennis KJ, Affek HP, Passey BH, Schrag DP, & Eiler JM (2011) Defining an absolute reference frame for 'clumped’ isotope studies of CO2. Geochim. Cosmochim. Acta 75(22):7117-7131.
Eiler JM (2011) Paleoclimate reconstruction using carbonate clumped isotope thermometry. Quat. Sci. Rev. 30(25-26):3575-3588.
Eagle RA, Eiler JM, Tripati AK, Ries JB, Freitas PS, Hiebenthal C, Wannamaker AD, Traviani M, Elliot M, Marenssi S, Nakamura K, Ramirez P, & Roy K (2013) The influence of temperature and seawater carbonate saturation state on 13C–18O bond ordering in bivalve mollusks. Biogeosciences 10(7):4591-4606.
Ghosh P, Adkins J, Affek H, Balta B, Guo WF, Schauble EA, Shrag D, & Eiler JM (2006) 13C-18O bonds in carbonate minerals: A new kind of paleothermometer. Geochim. Cosmochim. Acta 70(6):1439-1456.
Guo WF, Mosenfelder JL, Goddard WA, & Eiler JM (2009) Isotopic fractionations associated with phosphoric acid digestion of carbonate minerals: Insights from first-principles theoretical modeling and clumped isotope measurements. Geochim. Cosmochim. Acta 73(24):7203-7225.
Halevy I, Fischer WW, & Eiler JM (2011) Carbonates in the Martian meteorite Allan Hills 84001 formed at 18 +/- 4 degrees C in a near-surface aqueous environment. Proceedings of the National Academy of Sciences of the United States of America 108(41):16895-16899.
Pierrehumbert RT, Abbot DS, Voigt A, & Koll D (2011) Climate of the Neoproterozoic. Annual Review of Earth and Planetary Sciences, Vol 39, Annual Review of Earth and Planetary Sciences, eds Jeanloz R & Freeman KH), Vol 39, pp 417-460.
Saenger C, Affek H, Felis T, Thiagarajan N, Lough JM, & Holcomb M (2012) Carbonate clumped isotope variability in shallow water corals: Temperature dependence and growth-related vital effects. Geochim. Cosmochim. Acta 99(0):224-242.
Thiagarajan N, Adkins J, & Eiler J (2011) Carbonate clumped isotope thermometry of deep-sea corals and implications for vital effects. Geochim. Cosmochim. Acta 75(16):4416-4425.
Tripati AK, et al. (2010) 13C-18O isotope signatures and 'clumped isotope’ thermometry in foraminifera and coccoliths. Geochim. Cosmochim. Acta 74(20):5697-5717.
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Clark Johnson
Project Investigator
Huifang Xu
Co-Investigator
Brian Beard
Collaborator
John Eiler
Collaborator
Nicholas Levitt
Collaborator
-
RELATED OBJECTIVES:
Objective 2.1
Mars exploration.
Objective 4.1
Earth's early biosphere.
Objective 7.1
Biosignatures to be sought in Solar System materials
Objective 7.2
Biosignatures to be sought in nearby planetary systems