2013 Annual Science Report
 Massachusetts Institute of Technology
Reporting | SEP 2012 – AUG 2013
Massachusetts Institute of Technology
Reporting | SEP 2012 – AUG 2013
Early Animals: The Genomic Origins of Morphological Complexity
Project Summary
Understanding the origins of life’s complexity here on Earth is paramount to finding it elsewhere in the universe. The fossil record indicates that complexity on Earth arose in a near geological moment—the famous Cambrian explosion—about 525 million years ago. However, molecular sequence analyses indicate that complex animals actually arose nearly 200 million years before they make their first appearance in the fossil record (Erwin et al. 2011). This disparity between the advent of morphological complexity and its appearance in the fossil record motivates an interesting question: why is it that we cannot detect complex life here on Earth for nearly 200 million years? And if we cannot detect it on Earth, what hope would we have on another distant Earth-like planet? Our research is focused on addressing this question by trying to obtain a better understanding of what encodes morphological complexity in the genome. Our research (Heimberg et al. 2008; Philippe et al. 2001; Tarver et al. 2013) suggests that a group of non-coding RNA genes—microRNAs—might be instrumental for the advent and maintenance of complexity in animals, and therefore sequencing the genomes and the transcriptomes (the expressed component of the genome) from carefully chosen taxa might allow us to better understand the biology of animals that predated the Cambrian explosion.
Project Progress
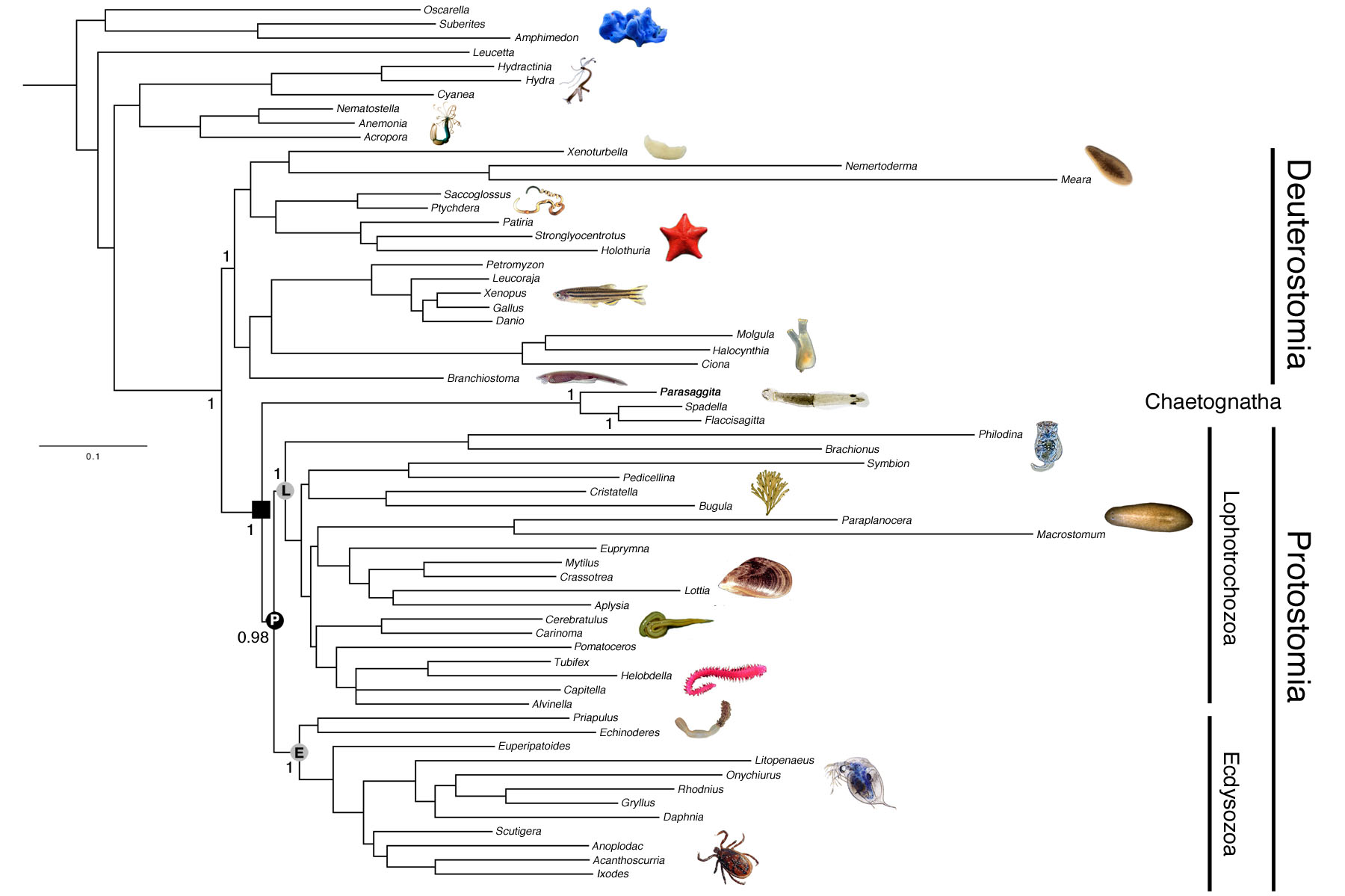
To date, we have sequenced the genome and both the mRNA and miRNA transcriptomes of the chaetognath Parasaggita elegans. Chaeotognaths are relatively complex animals and are the first predators that make their appearance in the fossil record during the Early Cambrian (Vannier et al. 2007). The genome assembly of Parasagitta consists of 866,422 scaffolds with the longest scaffold being 28,090 bp. Our assembled genome is 1.2 Gb, and our calculated N50 value is 750 bp. The mRNA transcriptome assembly of is 44Mb with an N50 of 1738bp. For the small RNA transcriptome, we analyzed 93,829 non-redundant sequences, all of which were expressed four or more times in our library. Using both genomic and mRNA transcriptome sequence we compiled a data set of the amino acid sequences from 186 protein-coding genes, and aligned them to an existing phylogenomics data set (Philippe et al., 2011). Our phylogenetic analysis (Figure 1) strongly suggests that chaetognaths are basal to the two major protostome clades, the lophotrochozoans and the ecydysozoans, similar to what others have recently proposed (e.g., Marlétaz et al., 2008). This phylogenetic position indicates that the chaetognath lineage split from the main protostome lineage ~650 Ma, over 100 Ma before they make their appearance in the fossil record. Our analysis of the microRNAs (Tarver et al. 2013) is consistent with this phylogenetic position as the chaetognath P. elegans shares nine miRNAs with ecdysozoans and lophotrochozoans not present in deuterostomes or in more basal organisms like sponges and jellyfish. However, the lophotrochozoans and ecydysozoans also possess three additional miRNAs not present in either our small RNA library or in the genome of the chaetognath, miR-36, miR-67 and miR-317 (Figure 2), suggesting that these miRNAs evolved in the protostome lineage after the lineage split between chaetognaths and the traditional protostomes.
Because the phylogenetic position of chaetognaths appears to be (finally!) firmly established we can now properly evaluate the taxon’s relative microRNA repertoire. Our data indicate that the chaetognath has lost very few miRNAs – only miR-242 and miR-2001 appear to be secondarily lost, consistent with the observation that chaetognaths are relatively complex animals with no obvious signs of secondary simplification, highlighting the disparity between the origins of morphological complexity versus its manifestation in the fossil record.

Publications
-
Laflamme, M., Darroch, S. A. F., Tweedt, S. M., Peterson, K. J., & Erwin, D. H. (2013). The end of the Ediacara biota: Extinction, biotic replacement, or Cheshire Cat?. Gondwana Research, 23(2), 558–573. doi:10.1016/j.gr.2012.11.004
-
Peterson, K. J., Su, Y-H., Arnone, M. I., Swalla, B., & King, B. L. (2013). MicroRNAs support the monophyly of enteropneust hemichordates. J. Exp. Zool. (Mol. Dev. Evol.), 320(6), 368–374. doi:10.1002/jez.b.22510
-
Robinson, J. M., Sperling, E. A., Bergum, B., Adamski, M., Nichols, S. A., Adamska, M., & Peterson, K. J. (2013). The Identification of MicroRNAs in Calcisponges: Independent Evolution of MicroRNAs in Basal Metazoans. J. Exp. Zool. (Mol. Dev. Evol.), 320(2), 84–93. doi:10.1002/jez.b.22485
-
Tarver, J. E., Sperling, E. A., Nailor, A., Heimberg, A. M., Robinson, J. M., King, B. L., … Peterson, K. J. (2013). miRNAs: Small Genes with Big Potential in Metazoan Phylogenetics. Molecular Biology and Evolution, 30(11), 2369–2382. doi:10.1093/molbev/mst133
- Peterson, K.J. (2013). Origin and Early Evolution of Animals. In: Losos, J. (Eds.). Oxford Bibliographies in Evolutionary Biology. New York: Oxford University Press.
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Ben King
Co-Investigator
Kevin Peterson
Co-Investigator
Davide Pisani
Co-Investigator
Albert Poustka
Co-Investigator
Andreas Schmidt-Rhaesa
Co-Investigator
-
RELATED OBJECTIVES:
Objective 4.1
Earth's early biosphere.
Objective 4.2
Production of complex life.