2013 Annual Science Report
 Carnegie Institution of Washington
Reporting | SEP 2012 – AUG 2013
Carnegie Institution of Washington
Reporting | SEP 2012 – AUG 2013
Project 3: The Origin, Evolution, and Volatile Inventories of Terrestrial Planets
Project Summary
We study the origin and evolution of the terrestrial planets with a special emphasis on CHON volatiles, their delivery and retention in the deep interiors of terrestrial planets. We will experimentally investigate how CHON volatiles may be retained even during magma ocean phases of terrestrial evolution. We investigate the early Earth’s recycling processes studying the isotopic composition of diamonds, diamond inclusions, and associated lithologies. We continue to integrate new information from the NASA Messenger Mission to Mercury into the broader context of understanding the inner Solar System planets.
Project Progress
Project 3. The Origin, Evolution, and Volatile Inventories of Terrestrial Planets
3.1 Speciation of C-O-H-N Fluids at high pressures and temperatures
The research focus of CoI Mysen and collaborator Foustoukos during this year has been structural, compositional, and isotopic characterization of fluids and melts in silicate-COHN systems to several GPa and about 1000˚C. All experiments were conducted with externally-heated hydrothermal diamond anvil cells (HDAC).
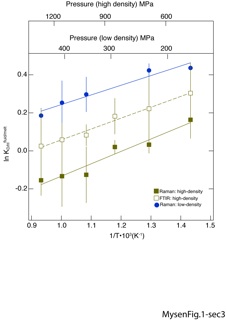
Speciation of C-O-H-N volatiles in alkali aluminosilicate melts and fluids and of silicate dissolved in C-O-H-N fluid has been determined in-situ to 900˚C and > 5 GPa under reducing (near IW) and oxidizing redox conditions. Under oxidizing conditions, molecular N2 is the only N-bearing species in fluids and melts. Under reducing conditions, molecular NH3 and ammine groups, NH2-, were detected in fluids and melts with NH3/NH2 ratio varying between 0.15 and 0.75 in the 425˚-800˚C temperature. For the NH2,NH3 exchange equilibrium the enthalpy change, ∆H, equals 19±8 kJ/mol and 61±9 kJ/mol for melt and fluid, respectively. The fluid/melt partition coefficient, (NH3 + NH2-)fluid/(NH3 + NH2-)melt, ranges from 8 to 3 with ∆H=45±12 kJ/mol. The ∆H of hydrogen bonding among the N-H groups in fluids is 28.4±4.3 kJ/mol. Hydrogen bonding can be detected at 650˚C and below. This behavior resembles that of H2O at similar temperatures and pressures. For reduced carbon species CH4 and CH3 groups exist in melt with the methyl groups likely bonded to Si. Raman bands assigned to C-H stretching in CHxDy isotopologues and CH4 groups (including CH3) were employed to determine the CH4/CHxDy ratio in fluids and melts. This ratio decreases from 1.5-2 at 500˚C to between 1.2 and 1 with 800˚C with ∆H-values of 13.6±2.1 and 5.5±1.1 kJ/mol for melt and fluid, respectively. The CH4/CHxDy fluid/melt partition coefficient ranges between ~16 and ~3 with ∆H=33±6 kJ/mol. This behavior of deuterated and protonated complexes is ascribed to speciation of volatile and silicate components in fluids and melts in a manner that is conceptually similar to D/H partitioning among complexes and phases in brines and hydrous silicate systems.
In the compositionally simplest system, H2-D2-D2O-H2O buffered with the Ti-TiO2 reaction, Raman spectroscopy was employed to explore the relative distribution of hydrogen and deuterium isotopologues of the H2 and H2O in supercritical fluids in the 300˚-800˚C and 0.3-1.3 GPa temperature and pressure range, respectively. An important finding was that estimated enthalpy for the H2(aq)-D2(aq) disproportionation reaction (DHrxn) is about -3.4 kcal/mol, which differs greatly from the +0.16 kcal/mol predicted for the exchange reaction in the gas phase by statistical mechanics models. The negative DHrxn values for the H2(aq)-D2(aq)-HD(aq) equilibrium and the apparent decrease of the equilibrium constant with increasing temperature are possibly triggered by differences of the Henry’s Law constant between the H- and D-bearing species dissolved in supercritical aqueous solutions. Such effects are attributed to the stronger hydrogen bonding in the O-H∙∙∙O relative to the O-D∙∙∙O environment. The exothermic behavior of the exchange reaction would enhance the stability of H2 and D2 relative to HD. Accordingly, the significant energy difference of the internal H2(aq)-D2(aq)-HD(aq) equilibrium translates to strong differences of the fractionation effects between the H2O-H2 and D2O-D2[ isotope exchange relationships. The D/H isotope fractionation factors between H2O-H2(aq) and D2O-D2(aq) differ by 363 ‰ in the 600 – 800 oC temperature range, and are indicative of the greater effect of D2O contribution to the ∂D isotopic composition. The latter pilot study was expanded to exploration speciation of and D/H fractionation between species in COH fluids was explored in-situ in the HDAC via reactions in the CH4-D2O-H2O to 800˚C and 4.5 GPa by using similar methods. Under reducing conditions, CH4, H2, H2O and their deuterated isotopolgues were observed, the concentrations of which are in accord with values obtained by numerical simulations. In the study of D/H fractionation, there are discreet differences of the equilibrium constants describing the relationship between the CH3D-CH2D2-CHD3-CH4 species dissolved in supercritical water or in homogeneous gas phase. The bulk D/H methane composition in the liquid- system is twice that of the D/H molar ratios recorded in the gas-bearing system. Accordingly, condensed-phase isotope effects are inferred to play a key role on the evolution of H/D isotopologues, likely induced by differences in the solubility of the isotopic molecules driven by the excess energy/entropy developed during mixing of non-polar species in the H2O-D2O structure. Our experiments show that isotope fractionation effects need to account for the presence of condensed matter (e.g. melts, magmatic fluids), even at conditions at which theoretical models suggest minimal (or nonexistent) isotope exchange, but comparable to those of the Earth’s interior.
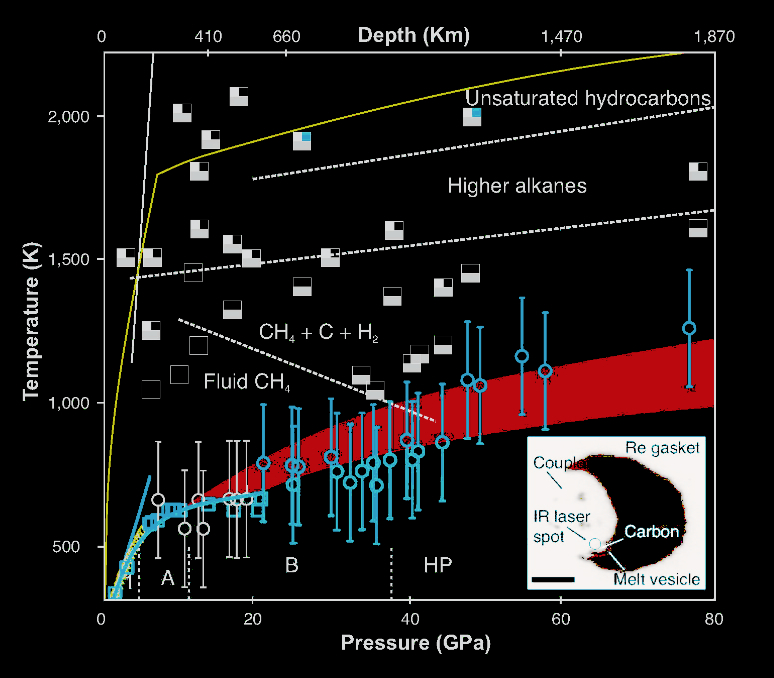
3.2 Carbon precipitation from heavy hydrocarbon fluid in deep planetary interiors
CoI Goncharov has been focusing on reactions of carbonaceous volatiles in deep planetary interiors. The phase diagram of C-H system is of great importance to planetary sciences, as hydrocarbons comprise a significant part of icy giant planets and are involved in reduced C-O-H fluid in deep Earth. Here we use resistively and laser-heated diamond anvil cells to measure methane melting and chemical reactivity up to 80 GPa and 2000 K. We show that methane melts congruently below 40 GPa. Hydrogen and elementary carbon appear at temperatures above 1200 K, whereas heavier alkanes and unsaturated hydrocarbons (>24 GPa) form in melts above 1500 K. The phase composition of C-H fluid evolves toward heavy hydrocarbons at pressures and temperatures representative of Earth’s lower mantle. We argue that reduced mantle fluids precipitate diamond upon re-equilibration to lighter species in the upwelling mantle. Likewise, our findings suggest that geophysical models of Uranus and Neptune require reassessment because chemical reactivity of planetary ices is underestimated.
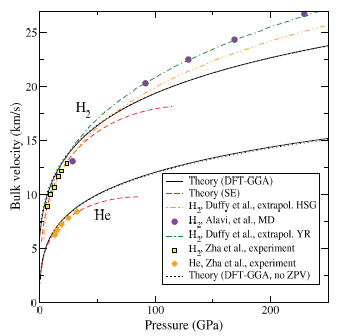
3.3 Sound velocities of hexagonal close-packed H2 and He under pressure
CoI Goncharov and collaborators have been studying hydrogen at extreme pressures. The bulk and shear sound velocities in solid H2 and He calculated with the SE many-body intermolecular potential are in good agreement with experiment. The results can serve as a baseline for planetary and astrophysical models and provide a basis for extrapolation to more extreme conditions.
3.4 Bonding, structures, and band gap closure of hydrogen at high pressures
CoI Goncharov and collaborators have studied dense hydrogen and deuterium experimentally up to 320 GPa and using ab initio molecular dynamic (MD) simulations up to 370 GPa between 250 and 300 K. Raman and optical absorption spectra show significant anharmonic and quantum effects in mixed atomic and molecular dense phase-IV of hydrogen. In agreement with these observations, ab initio MD simulations near 300 K show extremely large atomic motions, which include molecular rotations, hopping, and even pair fluctuations, suggesting that phase IV may not have a well-defined crystalline structure. The structurally diverse layers (molecular and grapheme like) are strongly
coupled, thus opening an indirect band gap; moreover, at 300 GPa, we find fast synchronized intralayer structural fluctuations. At 370 GPa, the mixed structure collapses to form a metallic molecular Cmca-4 phase, which exhibits a new interstitial valence charge bonding scheme.
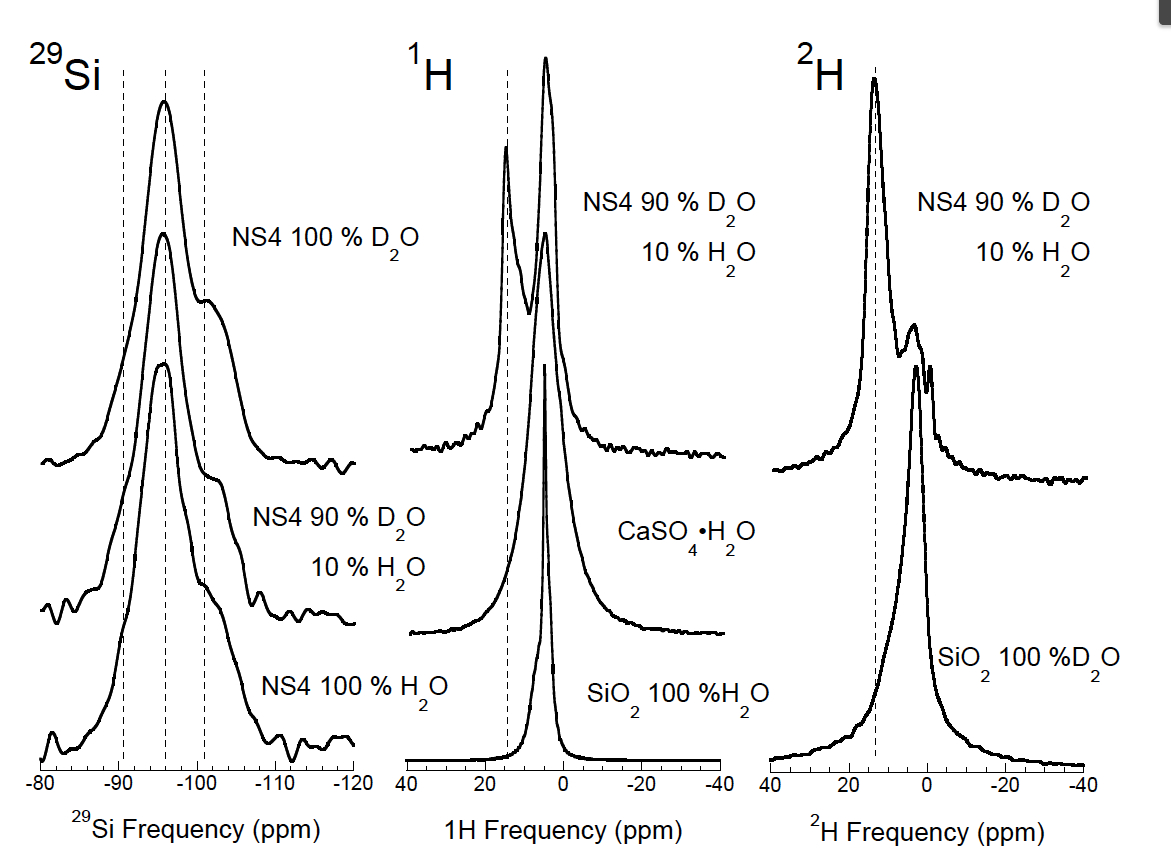
3.5 Measuring intramolecular isotope partitioning in silicate glasses quenched from melts via Nuclear Magnetic Resonance Spectroscopy
Postdoctoral fellow Ying Wang working with PI Cody, CoI Mysen, and collaborator Foustoukos have been exploring intramolecular D-H fractionation in melts quenched to glasses using D (2H) and 1H solid state Nuclear Magnetic Resonance (NMR) Spectroscopy. The solution mechanism by which water interacts with silicate melts is very complex, lending to the potential for interesting and surprising isotopic behavior during magma devolalitization. A particularly interesting period in Earth’s history was the primary devolatization during crystallization of the magma ocean phase(s), both after primary accretion and again during the Moon forming impact. It is currently known that water in the upper mantle is depleted in deuterium relative to the oceans and there exists evidence that the D/H ratio of the Earth’s oceans may have evolved through time. There is no clearly defined mechanism to explain these observations. Classically one does not expect any detectible D/H fractionation at the high temperatures that typify magma systems (typically in excess of 1000 °C), however, we find enormous D/H intramolecular fractionation between different molecular environments within silicate glasses quenched from melts synthesized under upper mantle conditions of T (1400 °C) and P (20000 Bar). Through experiment and simulation we rule out kinetic isotope effects and conclude that this fractionation results from molar volume isotope fractionation. This first investigation is in review in Earth Planetary Science Letters. We are exploring this system in across a broader spectrum of melt compositions to gain better understanding of the detailed fractionation mechanisms.
3.6 Investigations into water in diamond inclusions
Diamonds and the minerals that they include carry the deepest, oldest samples from Earth’s mantle that are available for study. CoI’s Shirey and Hauri have looked at mineral inclusions in diamonds from Earth’s mantle transition zone, thought to have originated from depths between 410 and 660 km. A goal of this project is to understand the extent to which volatiles, such as water, are transported into Earth’s deep interior. This process, widespread on Earth, could be typical of the type of volatile introduction that occurs on other rocky planets if they were to have tectonics akin to plate tectonics.
The analyses have been conducted on polished diamonds from the Juina 5 and Collier 4 kimberlite pipes in the Juina kimberlite field, Brazil using the DTM ion probe, a NanoSIMS 50L. This is an ongoing study for which only preliminary data are available. Nonetheless the data suggests diamonds remain closed and there is not pervasive loss of water from the inclusion to the diamond interface. Some first order conclusions can be drawn. The lowest water content phases (ol, maj, cpx) are in the low range for oceanic mantle estimates of 100-200 ppm H2O. Stishovite is undersaturated with respect to water at its recombined Al content. Majorite is variable, undersaturated, and still carries significant water while Ca-Si-Ti-perovskite is very water-rich. The phases measured here for water content are associated with diamonds that have light δ13C and would support the introduction of this water by slab subduction. The data supports a mantle model where the slab is wetter, the ambient mantle is drier, the Transition Zone (TZ) is not saturated, and/or diamonds form from melts/fluids with low aH2O.
3.7 Understanding the origins of the Inner Planets- The Study of Mercury
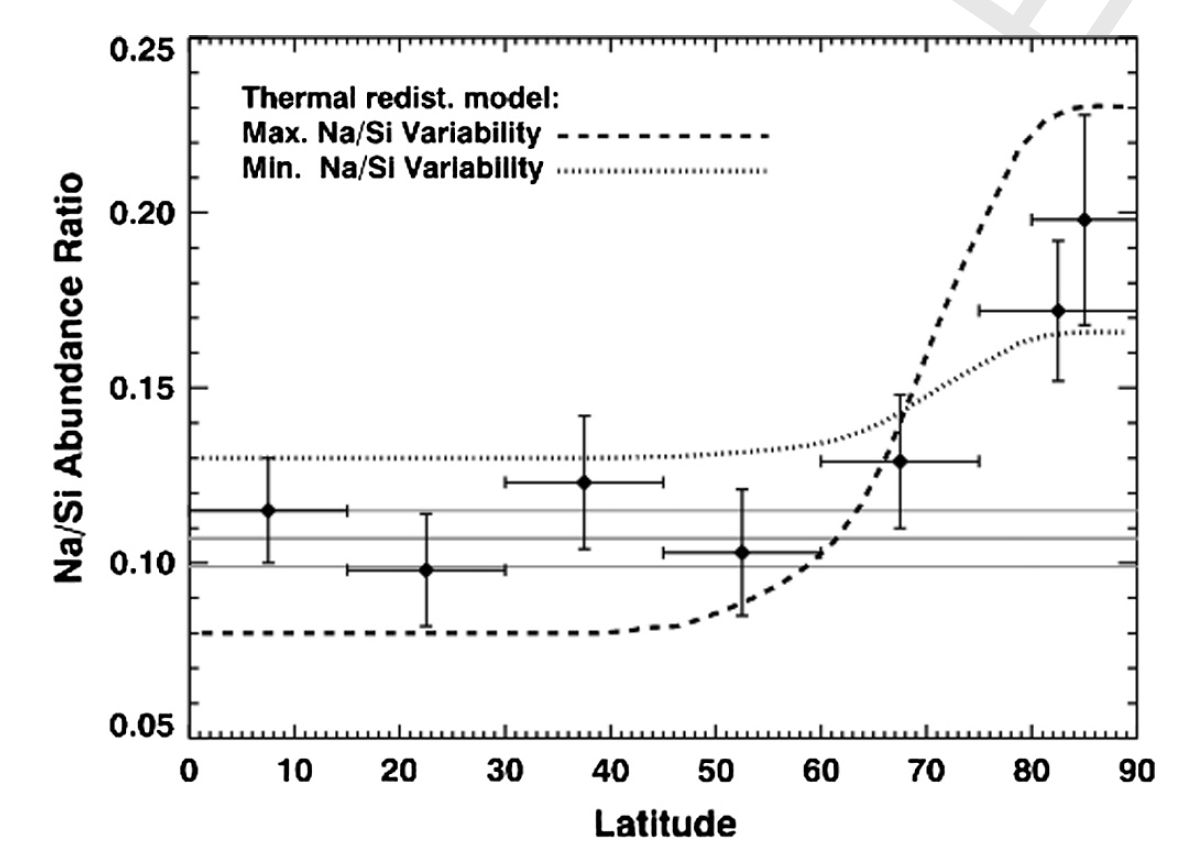
Co-I Solomon is the Principal Investigator and Co-I Nittler the Deputy Principal Investigator of the MESSENGER mission to Mercury. MESSENGER, now in its second extended mission, has continued in the last year to return a wealth of data from its payload of scientific instruments. Three independent lines of evidence – neutron flux, surface reflectance and MESSENGER-informed thermal modeling – all led to the conclusion that there are large deposits of water ice sited in regions of permanent shadow within polar impact craters. Moreover, several of these deposits most likely are overlain by deposits of organic matter. The stability over geological timescales of water and organics on the innermost planet confirms the ubiquity of these astrobiologically important materials throughout the solar system. Continued chemical mapping of the planet has revealed both major-element heterogeneity correlated with crustal thickness and a surprisingly high and variable abundance of the volatile element sodium on Mercury. The latter provides further evidence that Mercury contains a similar inventory of volatile elements to other terrestrial planets despite its much higher density and apparently highly reduced chemistry.

Publications
-
Foustoukos, D. I., & Mysen, B. O. (2012). D/H fractionation in the H2–H2O system at supercritical water conditions: Compositional and hydrogen bonding effects. Geochimica et Cosmochimica Acta, 86, 88–102. doi:10.1016/j.gca.2012.03.003
-
Foustoukos, D. I., & Mysen, B. O. (2013). H/D methane isotopologues dissolved in magmatic fluids: Stable hydrogen isotope fractionations in the Earth’s interior. American Mineralogist, 98(5-6), 946–954. doi:10.2138/am.2013.4419
-
Freiman, Y. A., Grechnev, A., Tretyak, S. M., Goncharov, A. F., & Hemley, R. J. (2013). Sound velocities in solid hydrogen under pressure. Low Temp. Phys., 39(5), 423. doi:10.1063/1.4807043
-
Goncharov, A. F., Tse, J. S., Wang, H., Yang, J., Struzhkin, V. V., Howie, R. T., & Gregoryanz, E. (2013). Bonding, structures, and band gap closure of hydrogen at high pressures. Physical Review B, 87(2), None. doi:10.1103/physrevb.87.024101
-
Lobanov, S. S., Chen, P-N., Chen, X-J., Zha, C-S., Litasov, K. D., Mao, H-K., & Goncharov, A. F. (2013). Carbon precipitation from heavy hydrocarbon fluid in deep planetary interiors. Nat Comms, 4. doi:10.1038/ncomms3446
-
Mysen, B. (2013). Effects of fluid and melt density and structure on high-pressure and high-temperature experimental studies of hydrogen isotope partitioning between coexisting melt and aqueous fluid. American Mineralogist, 98(10), 1754–1764. doi:10.2138/am.2013.4449
-
Mysen, B. (2013). Hydrogen isotope fractionation between coexisting hydrous melt and silicate-saturated aqueous fluid: An experimental study in situ at high pressure and temperature. American Mineralogist, 98(2-3), 376–386. doi:10.2138/am.2013.4247
-
Mysen, B. (2013). Structure–property relationships of COHN-saturated silicate melt coexisting with COHN fluid: A review of in-situ, high-temperature, high-pressure experiments. Chemical Geology, 346, 113–124. doi:10.1016/j.chemgeo.2012.10.006
-
Mysen, B. O. (2012). Silicate-COH melt and fluid structure, their physicochemical properties, and partitioning of nominally refractory oxides between melts and fluids. Lithos, 148, 228–246. doi:10.1016/j.lithos.2012.06.005
-
Mysen, B. O., Mibe, K., Chou, I-M., & Bassett, W. A. (2013). Structure and equilibria among silicate species in aqueous fluids in the upper mantle: Experimental SiO2-H2O and MgO-SiO2-H2O data recorded in situ to 900°C and 5.4 GPa. Journal of Geophysical Research: Solid Earth, 118(12), 6076–6085. doi:10.1002/2013jb010537
-
Mysen, B. O., Tomita, T., Ohtani, E., & Suzuki, A. (2014). Speciation of and D/H partitioning between fluids and melts in silicate-D-O-H-C-N systems determined in-situ at upper mantle temperatures, pressures, and redox conditions. American Mineralogist, 99(4), 578–588. doi:10.2138/am.2014.4575
-
Roskosz, M., Bouhifd, M. A., Jephcoat, A. P., Marty, B., & Mysen, B. O. (2013). Nitrogen solubility in molten metal and silicate at high pressure and temperature. Geochimica et Cosmochimica Acta, 121, 15–28. doi:10.1016/j.gca.2013.07.007
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Celia Dalou
Co-Investigator
Marilyn Fogel
Co-Investigator
Dionysis Foustoukos
Co-Investigator
Erik Hauri
Co-Investigator
Yoko Kebukawa
Co-Investigator
Ying Wang
Co-Investigator
-
RELATED OBJECTIVES:
Objective 1.1
Formation and evolution of habitable planets.
Objective 3.1
Sources of prebiotic materials and catalysts
Objective 4.1
Earth's early biosphere.







