2012 Annual Science Report
 University of Hawaii, Manoa
Reporting | SEP 2011 – AUG 2012
University of Hawaii, Manoa
Reporting | SEP 2011 – AUG 2012
Amino Acid Alphabet Evolution
Project Summary
A genetically encoded alphabet of just 20 amino acids has produced the universe of protein structures and functions found throughout Earth’s biosphere. Relationships within this amino acid alphabet are responsible for fundamental biological phenomena, such as protein folding and patterns of molecular evolution. In attempting to unravel these relationships, considerable scientific ingenuity has been spent developing systems to simplify the genetically encoded alphabet of 20 amino acids while minimizing the associated loss of chemical diversity. These efforts present an opportunity to generate a composite picture of the properties that link the amino acids as a set. We are therefore investigating whether different simplification schemes (“simplified amino acid alphabets”), including those derived from very different approaches, can be combined to create a coherent description of amino acid similarity. By understanding the organization and relationships between amino acids on Earth, we hope to shed light on the chemical logic to be expected as a product of evolution in extraterrestrial environments.
An extensive scientific literature has converged on surprisingly clear agreement that a subset of only around half of the 20 genetically encoded amino acids was likely present from the inception of genetic coding (the “early” amino acids), and an equal sized subset was incorporated through subsequent evolution (the “late” amino acids). A further widespread assumption is that, as the set expanded, natural selection favored the addition of amino acids that extended the range of protein structures and functions. We initiated a quantitative investigation for consilience between these two important ideas.
Project Progress
Deconstructing Amino Acid Similarity
We have developed a new method for the objective comparison of multiple simplified amino acid alphabets, generated by any means. We have applied this method to an all against all comparison of 34 simplification schemes reported within previous scientific literature. This study revealed that different simplification schemes did not cluster according to the approach by which they were calculated. To understand why this should be so, we extended our alphabet comparison methods to produce a detailed view of amino acid similarity according to the consensus of two or more simplified alphabets. We used this calculation to show a strong and unanticipated match between the consensus view of all “top down” approaches (which derive their measurements of amino acid similarity by considering the roles that amino acids play within proteins and protein evolution) and “bottom up” approaches (which derive their measurements of amino acid similarity from the physics and chemistry of individual amino acid molecules). Taken together, these results indicate that although individual simplified alphabets differ from one another and continue to emerge, taken as a whole they can be deconstructed to reveal a coherent and meaningful picture of amino acid similarity. Our new understanding offers new tractability for fundamental questions from astrochemistry to synthetic biology.
Testing Ideas for the Evolution of the Genetically Encoded Amino Acid Alphabet
We developed a novel method to represent the protein-building potential of a set of amino acids in terms of the area(s) of chemistry space that these molecules populate. Using this method, we were able to conduct the first direct test of the adaptive hypothesis that the “early” amino acid alphabet grew through the addition of amino acids that enabled the construction of new protein structures and functions. Specifically, our analysis revealed remarkably strong support for the concept of adaptive growth of the amino acid alphabet in terms of amino acid size (Van der Waals volume) and hydrophobicity (LogP), producing a clear signal despite the simplicity of our test. However, amino acid charge (measured as pI) showed no such tendency. We discussed a range of interpretations, including the reasonable expectation that an optimal set of amino acid pI values might entail a more sophisticated distribution than our current test would detect. This work has been submitted for publication in the Journal of Systems Chemistry.
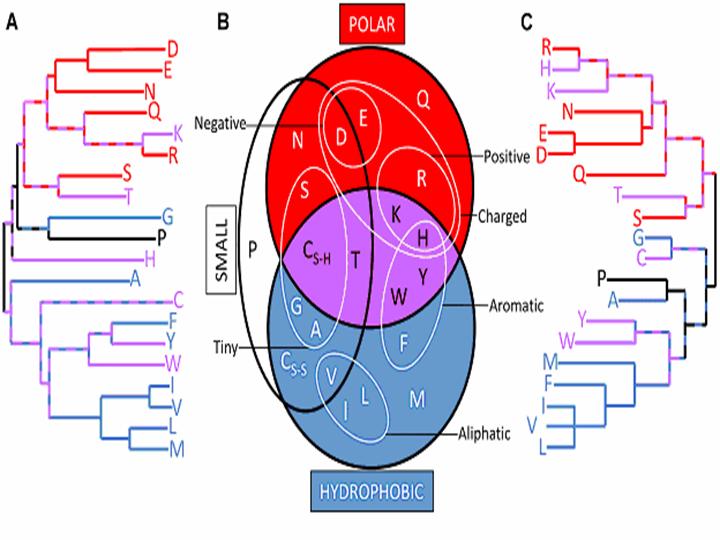
Amino acid similarity relationships derived by “top down” methods closely resemble those derived by “bottom up” approaches. Dendrogram constructed by neighbor-joining using the similarity data from 29 “top down” (A) and 5 “bottom up” (C) amino acid simplifications. Long branches indicate that an amino acid is rarely grouped with any other as part of a simplifi-cation scheme. Short path lengths be-tween amino acids suggest high similarity between them. Comparing both dendrograms with a redrawn version of a commonly used chemical property Venn diagram (B) uncovers the physicochemical basis for many of the dendrogram patterns. The hydro-phobic (blue), polar (red), and both hydrophobic and polar (purple) amino acids are colored to highlight the prin-cipal separation of the dendrograms. (Note: cysteine appears twice in (b) to reflect its role in disulfide bridges).
Publications
-
Ilardo, M. A., & Freeland, S. J. (2014). Testing for adaptive signatures of amino acid alphabet evolution using chemistry space. Journal of Systems Chemistry, 5(1), 1. doi:10.1186/1759-2208-5-1
- Ilardo, M. (2013, Submitted). Questioning the RNA world. Astrobiology.
- Stephenson, J. & Freeland, S. (2012, In Review). Deconstructing amino acid similarity. Plos One.
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Stephen Freeland
Project Investigator
James Stephenson
Postdoc
Melissa Ilardo
Graduate Student
-
RELATED OBJECTIVES:
Objective 3.2
Origins and evolution of functional biomolecules
Objective 4.1
Earth's early biosphere.
Objective 4.2
Production of complex life.
Objective 6.2
Adaptation and evolution of life beyond Earth
Objective 7.1
Biosignatures to be sought in Solar System materials
Objective 7.2
Biosignatures to be sought in nearby planetary systems