2012 Annual Science Report
 Massachusetts Institute of Technology
Reporting | SEP 2011 – AUG 2012
Massachusetts Institute of Technology
Reporting | SEP 2011 – AUG 2012
Metabolic Networks From Cells to Ecosystems
Project Summary
Members of the Segre’ group use systems biology approaches to study the complex network of metabolic reactions that allow microbial cells to survive and reproduce under varying environmental conditions. The resource allocation problem that underlies these fundamental processes changes dramatically when multiple cells can compete or cooperate with each other, for example through metabolic cross-feeding. Through mathematical models of microbial ecosystems and computer simulations of spatially structured cell populations, the Segre’ team aims at understanding the environmental conditions and evolutionary processes that favor the emergence of multicellular organization in living systems.
Project Progress
1. Genetic interaction, hierarchical modules and the evolution of biological complexity:
Much of the complexity of how genetic perturbations produce phenotypic outcomes has to do with the lack of independence, or epistasis, between such perturbations: the phenotypic effect of one perturbation depends, in general, on the genetic background of previously accumulated modifications, i.e., on the network of interactions with other perturbations. Both computational and experimental data on epistasis in S. cerevisiae have revealed that these genetic interaction networks are organized into modules associated with specific biological functions.
Part of our recent research has been focused on understanding how these genetic interaction networks relate to the modular architecture of biological systems. We have recently discovered that special types of modular organization (e.g. such that the fitness of an organism can be quantitatively partitioned into a benefit component and a cost component) give rise to negative epistasis between mutations affecting such modules. This finding has potential implications both for understanding the evolutionary advantage of recombination, and for explaining diminishing returns epistasis effects observed between mutations in experimentally evolved microbes.
The benefit-cost model can be extended, leading to a general expression for epistasis given an arbitrary functional dependence of fitness on other traits. This expression demonstrates how epistasis relative to fitness can emerge despite the absence of epistasis relative to lower level traits, leading to a formalization of the concept of independence between biological processes. Our results suggest that epistasis may be largely shaped by the pervasiveness of pleiotropic effects and modular organization in biological networks. The theory we developed may have broader relevance to any instance in which two biological compartments combine together to give rise to a larger joint multi-compartment system.
Related publications:
Hsuan-Chao Chiu, Christopher J. Marx, Daniel Segre’: Epistasis from functional dependence of fitness on underlying traits, Proceedings of the Royal Society B: Biological Sciences (2012), 279, 4156–4164.
Christopher Jacobs and Daniel Segre’: Organization principles in genetic interaction networks, Adv Exp Med Biol (2012) 751:53-78. (Special volume on Evolutionary Systems Biology, Orkun Soyer Ed.)
2. Understanding the regulatory and environmental control of respiration
The capacity of microorganisms to respond to variable external conditions requires a coordination of environment-sensing mechanisms and decision-making regulatory circuits. Here, we seek to understand the interplay between these two processes by combining high-throughput measurement of time-dependent mRNA profiles with a novel computational approach that searches for key genetic triggers of transcriptional changes. Our approach helped us understand the regulatory strategies of a respiratorily versatile bacterium with an important biogeochemical role, and promising practical applications, Shewanella oneidensis, in minimal and rich media. By comparing expression profiles across these two conditions, we unveiled components of the transcriptional program that depend mainly on the growth phase. Conversely, by integrating our time-dependent data with a previously available large compendium of static perturbation responses, we identified transcriptional changes that cannot be explained solely by internal network dynamics, but are rather triggered by specific genes acting as key mediators of an environment-dependent response. These transcriptional triggers include known and novel regulators that respond to carbon, nitrogen and oxygen limitation. Our analysis suggests a sequence of physiological responses, including a coupling between nitrogen depletion and glycogen storage, partially recapitulated through dynamic flux balance analysis, and experimentally confirmed by metabolite measurements. Our approach is broadly applicable to other systems.
Related publication
Qasim K. Beg*, Mattia Zampieri*, Niels Klitgord, Sara B. Collins, Claudio Altafini, Margrethe H. Serres, Daniel Segre’: Detection of transcriptional triggers in the dynamics of microbial growth: application to the respiratorily versatile bacterium Shewanella oneidensis. Nucleic Acids Research, (2012) 40 (15): 7132-7149. (*equally contributing authors)
3. Predicting metabolic cross-feeding in multicellular systems
As automated gene annotation pipelines, network gap-filling algorithms, and high throughput experimental methods improve, it will become gradually feasible to model the metabolism of any sequenced microbe using genome-scale flux balance models. Yet, some of the most fundamental properties of natural microbial ecosystems and proto-multi-cellular systems crucially depend on aspects that are beyond the stoichiometries of individual biochemical species. These include contact- or metabolite-mediated interactions between different microbes, dynamical changes of the environment, spatial structure of the underlying geography and evolutionary competition between distinct subpopulations.
We are developing COMETS (Computation Of Microbial Ecosystems in Time and Space), an open source, broadly applicable and user-friendly platform for performing spatially distributed time-dependent flux balance simulations of microbial ecosystems. By taking advantage of the computational efficiency of flux balance model calculations, we implement a spatially structured lattice of interacting metabolic subsystems. These subsystems represent a level of detail that is intermediate between a fine-grained single-cell modeling approach, and a global mean-field approximation. The COMETS simulation engine combines a modified version of dynamic flux balance analysis with a finite differences approximation of diffusion dynamics, to simultaneously track the spatio-temporal fate of multiple environmental molecules and microbial species. The COMETS platform has the capacity to bridge multiple spatial and temporal scales, making it possible to observe long term dynamics of microbial populations growing in a given environmental setting, based on constant updates of local nutrient availabilities and exchanges, and ultimately determined by the metabolic activity of individual microbial species. Once completed, COMETS could be used as a platform for modeling the growth of a single bacterial species in a Petri dish, the phenotypic differentiation in a biofilm, or the seasonality of microbial communities in a specific geographical setting.
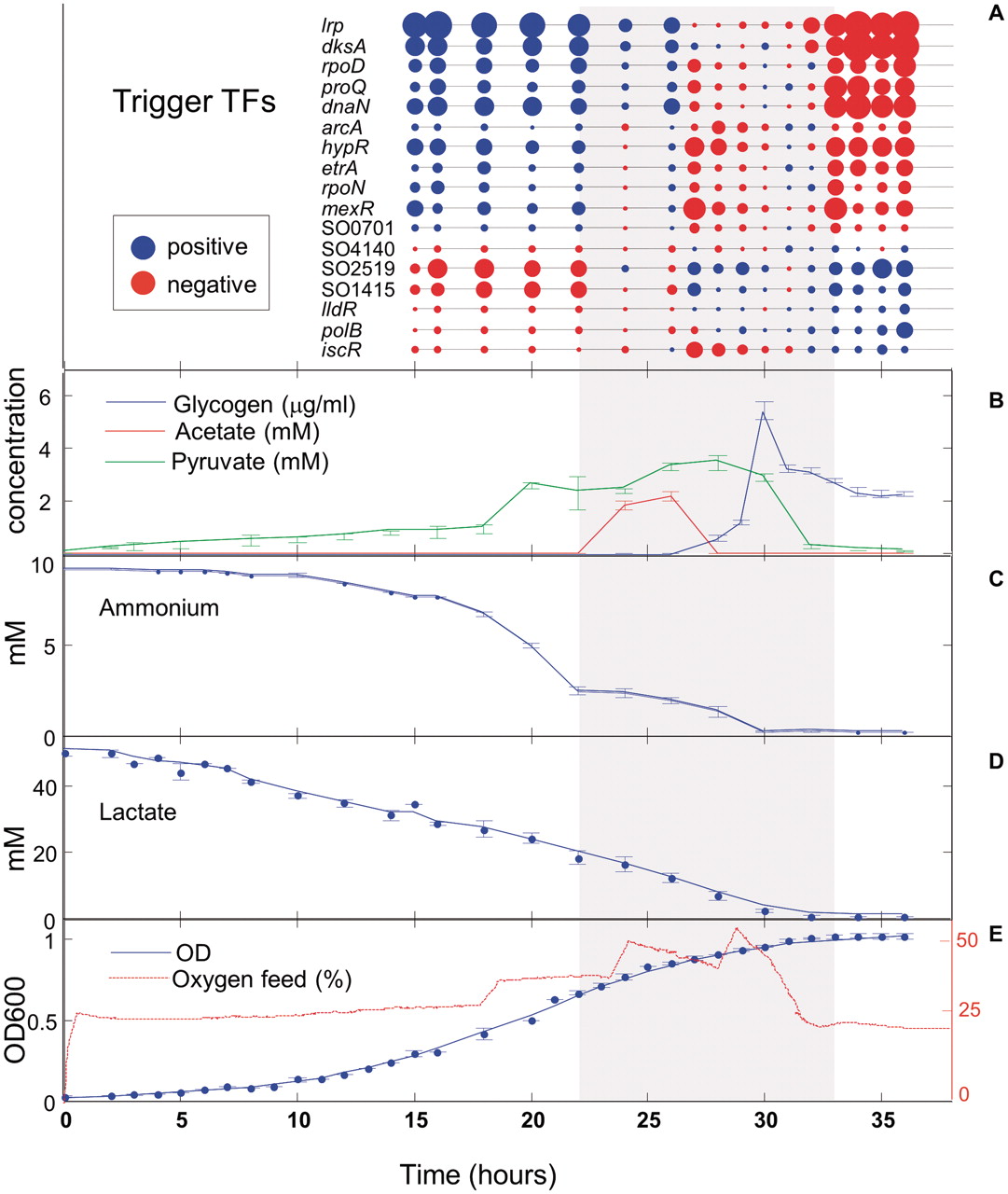
The figure illustrates the metabolic and transcriptional changes occurring during batch growth of Shewanella oneidensis in aerobic conditions.
Expression changes (A) over time of 17 key transcriptional regulators (red-low expression, blue-high expression). The size of circle represents level of expression. HPLC was used for quantifying acetate and pyruvate (B), whereas ammonium© was quantified using commercially available kit. Lactate (D) was quantified using HPLC and a commercially available kit. Optical density (E) of ∼1 ml culture samples was measured during growth of S. oneidensis at 600 nm using a spectrophotometer, and the external O2 feed data was collected using bioreactor software.
For more details, see Qasim K. Beg*, Mattia Zampieri*, Niels Klitgord, Sara B. Collins, Claudio Altafini, Margrethe H. Serres, Daniel Segre’: Detection of transcriptional triggers in the dynamics of microbial growth: application to the respiratorily versatile bacterium Shewanella oneidensis. Nucleic Acids Research, (2012) 40 (15): 7132-7149. (*equally contributing authors)
Publications
-
Beg, Q. K., Zampieri, M., Klitgord, N., Collins, S. B., Altafini, C., Serres, M. H., & Segre, D. (2012). Detection of transcriptional triggers in the dynamics of microbial growth: application to the respiratorily versatile bacterium Shewanella oneidensis. Nucleic Acids Research, 40(15), 7132–7149. doi:10.1093/nar/gks467
-
Chiu, H-C., Marx, C. J., & Segre, D. (2012). Epistasis from functional dependence of fitness on underlying traits. Proceedings of the Royal Society B: Biological Sciences, 279(1745), 4156–4164. doi:10.1098/rspb.2012.1449
-
Jacobs, C., & Segrè, D. (2012). Organization Principles in Genetic Interaction Networks. Advances in Experimental Medicine and Biology, None, 53–78. doi:10.1007/978-1-4614-3567-9_3
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Qasim Beg
Postdoc
Hsuan-Chao Chiu
Doctoral Student
Sara Collins
Doctoral Student
Christopher Jacobs
Doctoral Student
Niels Klitgord
Doctoral Student
-
RELATED OBJECTIVES:
Objective 3.4
Origins of cellularity and protobiological systems
Objective 4.2
Production of complex life.
Objective 5.2
Co-evolution of microbial communities
Objective 6.1
Effects of environmental changes on microbial ecosystems
