2010 Annual Science Report
 Massachusetts Institute of Technology
Reporting | SEP 2009 – AUG 2010
Massachusetts Institute of Technology
Reporting | SEP 2009 – AUG 2010
Metabolic Networks From Single Cells to Ecosystems
Project Summary
We use mathematical and computational approaches to study the dynamics and evolution of metabolism in individual microbes and in microbial ecosystems. In particular, we take advantage of sequenced genomes to study the complete network of biochemical reactions present in an organism. We have been extending these approaches from single genomes to multiple genomes, generating ecosystem-level models of metabolism, which can help us understand some of the key transitions in the history of life on our planet.
Project Progress
Sub-project 1: Predicting interactions in microbial ecosystems
In most natural environments, multiple microbial species interact with each other, forming complex ecosystems whose properties are poorly understood. In an effort to understand inter-microbial interactions, we have developed a computational method for predicting all possible symbiotic interactions between two given organisms across a large combinatorial set of possible minimal environmental conditions. Specifically, using the framework of flux balance analysis, we predicted environments in which either one or both microbes in a pair would not be able to grow without the other, inducing commensal (one-way) or mutualistic (two-way) interactions, respectively. Our algorithms can successfully recapitulate known inter-microbial interactions, and predict millions of new ones across any pair amongst different microbial species. Surprisingly, we find that it is always possible to identify conditions that induce mutualistic or commensal interactions between any two species. Hence, our method should help in mapping naturally occurring microbe-microbe interactions, and in engineering new ones through a novel, environment-driven branch of synthetic ecology.
Sub-project 2: Modeling the complex dynamics of enzyme-pathway coevolution
Metabolic pathways must have coevolved with the corresponding enzyme gene sequences. However, the evolutionary dynamics ensuing from the interplay between metabolic networks and genomes is still poorly understood. We have developed a computational model that generates putative evolutionary walks on the metabolic network using a parallel evolution of metabolic reactions with their catalyzing enzymes. Starting from an initial set of compounds and enzymes, we expanded the metabolic network iteratively by adding new enzymes with a probability that depends on their sequence-based similarity to already present enzymes. Thus, we obtain simulated time courses of chemical evolution in which we can monitor the appearance of new metabolites, enzyme sequences, or even entire organisms. We observe that new enzymes do not appear gradually but rather in clusters which correspond to enzyme classes. A comparison with Brownian motion dynamics indicates that our system displays biased random walks similar to diffusion on the metabolic network with long range correlations. This suggests that a quantitative molecular principle may underlie the concept of punctuated equilibrium as enzymes occur in bursts rather than by phyletic gradualism. Moreover, the simulated time courses lead to a putative time-order of enzyme and organism appearance. Among the patterns we detect in these evolutionary trends is a significant correlation between the time of appearance and their enzyme repertoire size. Hence, our approach to metabolic evolution may help understand the rise in complexity at the biochemical and genomic levels
Sub-project 3: Stability properties of autocatalytic metabolic cycles
We have investigated the stability properties of two different classes of metabolic cycles using a combination of analytical and computational methods. Specifically, we applied structural kinetic modeling (SKM) to a class of single input, single output metabolic cycles, and to a small autocatalytic cycle, and determined parameter regimes within which the cycles are very likely to be stable. We demonstrated that analytical methods can be used to understand the relationship between kinetic parameters and stability, and that results from these analytical methods can be confirmed with computational experiments. In addition, our results suggest that elevated metabolite concentrations and certain crucial saturation parameters can strongly affect the stability of the entire metabolic cycle. Our results suggest that evolutionary forces may select for metabolic network topologies with a high intrinsic probability of being stable. Furthermore, our conclusions support the hypothesis that certain types of metabolic cycles may have played a role in the development of primitive metabolism despite the absence of regulatory mechanisms.
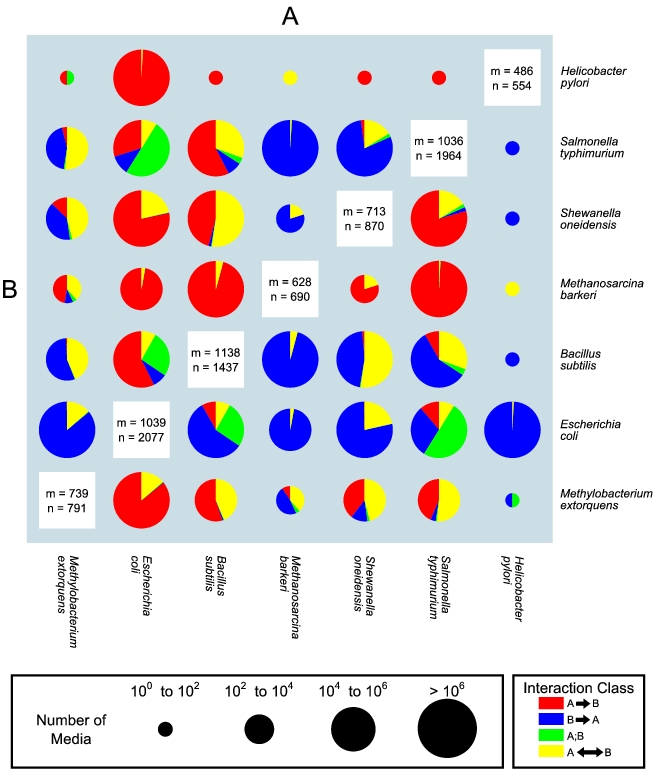
Predicted map of pairwise interactions between seven microbial species. The size of the pie chart is representative of the number of media that allow growth of both organisms, as defined in the pie chart size legend to the right. The relative amount of each interaction type is represented by the proportion of each wedge of the pie. Details on the numbers and types of interactions for each pair of organisms can be found in Table S1. The values of n and m reported in the white boxes on the diagonal indicate respectively the number of reactions and metabolites present in each stoichiometric model.
Publications
-
Klitgord, N., & Segrè, D. (2010). Environments that Induce Synthetic Microbial Ecosystems. PLoS Computational Biology, 6(11), e1001002. doi:10.1371/journal.pcbi.1001002
-
Reznik, E., & Segrè, D. (2010). On the stability of metabolic cycles. Journal of Theoretical Biology, 266(4), 536–549. doi:10.1016/j.jtbi.2010.07.023
-
Schütte, M., Skupin, A., Segrè, D., & Ebenhöh, O. (2010). Modeling the complex dynamics of enzyme-pathway coevolution. Chaos: An Interdisciplinary Journal of Nonlinear Science, 20(4), 045115. doi:10.1063/1.3530440
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Hsuan-Chao Chiu
Graduate Student
Niels Klitgord
Graduate Student
William Riehl
Graduate Student
-
RELATED OBJECTIVES:
Objective 4.1
Earth's early biosphere.
Objective 4.2
Production of complex life.
Objective 5.1
Environment-dependent, molecular evolution in microorganisms
Objective 5.2
Co-evolution of microbial communities
Objective 6.1
Effects of environmental changes on microbial ecosystems
