2011 Annual Science Report
 University of Wisconsin
Reporting | SEP 2010 – AUG 2011
University of Wisconsin
Reporting | SEP 2010 – AUG 2011
Project 4B: Iron Isotope Biosignatures - Synthesis of Abiotic and Biotic Laboratory Experiments
Project Summary
Experimental studies in geochemical systems analogous to ancient rock precursors are required to gain insight into the biogeochemical processes responsible for generating unique chemical or isotopic compositions in ancient rocks. We conducted a synthesis of prior and ongoing laboratory studies of Fe isotope fractionation during abiotic and biologically catalyzed (by dissimilatory microbial iron reduction, DIR) interaction between Fe(II) and Fe(III) oxide phases in the presence of dissolved silica, which was likely abundant in Precambrian oceans. In particular, previous studies of iron isotope fractionation during microbial reduction of an amorphous iron oxide-silica coprecipitate in high-silica, low-sulfate artificial Archean seawater were analyzed in combination with fractionation during abiotic Fe(II)-Fe(III) interaction to assess the rates and mechanisms of Fe isotope fractionation. The results show that although isotopic exchange rates were a function of solid and solution compositions, in all cases they were much higher than that determined in previous studies of aqueous Fe(III) and ferrihydrite interaction, highlighting the importance of electron exchange in promoting Fe atom exchange. When compared to analogous microbial reduction experiments with overlapping Fe(II) to Fe(III) ratios, isotopic exchange rates are faster in the biological experiments, likely due to promotion of atom exchange by the solid-phase Fe(II) produced during DIR. These findings provide constraints on the range of equilibrium iron isotope fractionations that may be produced by different geochemical processes. In addition, when interpreted in a Fe mass-balance context, our findings provide strong support for DIR as a mechanism for producing Fe isotope variations observed in Neoarchean and Paleoproterozoic marine sedimentary rocks.
Project Progress
Precambrian marine sedimentary rocks that formed at low temperatures, including shales, carbonates, and banded iron formations, contain the largest inventories of isotopically fractionated Fe in the Earth’s crust (e.g., ANBAR and ROUXEL, 2007; JOHNSON et al., 2008b). Redox processes are known to produce large Fe isotope fractionations at low temperatures in modern marine and fresh-water environments (e.g., BERGQUIST and BOYLE, 2006; FEHR et al., 2008; SEVERMANN et al., 2010; TEUTSCH et al., 2009), which suggests that redox changes were likely responsible for Fe isotope fractionation in Precambrian marine environments. Amorphous Fe(III) oxide (Fe(III)am) is one of the most important components of the Fe cycle in sedimentary environments (CORNELL and SCHWERTMANN, 2003). Silica has been used successfully to prevent the phase transformation of Fe(III)am in the presence of Fe(II), either in the dissolved form or synthesized together with Fe(III)am (WU et al., 2011a). Equilibrium Fe(II)-Fe(III)am 56Fe/54Fe fractionation factors of ‒3.17 ± 0.08 (2σ)‰ and ‒2.58 ± 0.14 (2σ)‰ have been obtained for Fe(III)am plus silica and the Fe(III)-Si coprecipitate (referred to hereafter as Fe-Si CP), respectively (WU et al., 2011a). The true equilibrium Fe(II)-Fe(III)am 56Fe/54Fe fractionation factor in the absence of silica has been inferred to be ~ ‒3.2 ‰ (WU et al., 2011a).
Microbial dissimilatory iron reduction (DIR) has been proposed to play a significant role in producing Fe isotope fractionations recorded in both modern and ancient marine environments (e.g., BEARD et al., 2003; JOHNSON et al., 2008a; STAUBWASSER et al., 2006; TANGALOS et al., 2010). Coupled electron and atom exchange has been shown to be the mechanism for Fe isotope fractionation during microbial dissimilatory iron reduction (CROSBY et al., 2007; PERCAK-DENNETT et al., 2011; WU et al., 2009), as well as abiological interaction between Fe(II)aq and various Fe(III) oxides (BEARD et al., 2010; WU et al., 2011a). A recent study investigated Fe isotope fractionation produced by DIR under conditions that simulated key aspects of Archean marine conditions, through use of an artificial seawater medium that contained dissolved silica and an amorphous Fe(III) oxide-silica coprecipitate designed to mimic the electron acceptors that likely existed in Archean ocean sediments (PERCAK-DENNETT et al., 2011). Microbial reduction of Fe-Si CP produced large quantities of aqueous, sorbed and solid-phase Fe(II) that had low-δ56Fe, providing strong support for DIR as an efficient means for producing the Fe isotope variations observed in the ancient rock record (PERCAK-DENNETT et al., 2011).
This study examined Fe isotope fractionation between Fe(II)aq and Fe-Si CPs under simulated Archean seawater conditions, and undertook a synthesis of these results with previous studies (PERCAK-DENNETT et al., 2011; WU et al., 2011a). The aim was to investigate Fe isotope exchange and fractionation in an abiological system analogous to the DIR system studied by Percak-Dennett et al. (2011). A three-isotope method was employed to determine the equilibrium Fe isotope fractionation factor that would be achieved during complete isotope exchange (e.g., BEARD et al., 2010; MATSUHISA et al., 1978). In addition, we explored the effects of Fe:Si molar ratio of the oxyhydroxide on equilibrium Fe isotope fractionation factors.
The results showed that although isotopic exchange rates were a function of solid and solution compositions, in all cases they were much higher than that determined in previous studies of aqueous Fe(III) and ferrihydrite interaction, highlighting the importance of electron exchange in promoting Fe atom exchange.
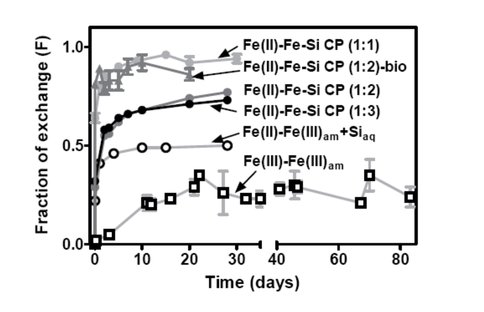
Comparison of isotopic exchange kinetics for experiments with different Fe(III)am phases. The approximate Fe:Si ratio of Fe-Si coprecipitates is shown in parenthesis. Circles represent abiological experiments, and triangles represent results from the dissimilatory Fe(III) reduction experiments reported in Percak-Dennett et al. (2011).
When compared to analogous microbial reduction experiments with overlapping Fe(II) to Fe(III) ratios, isotopic exchange rates were faster in the biological experiments, likely due to promotion of atom exchange by the solid-phase Fe(II) produced during DIR.
The equilibrium isotope fractionation (in 56Fe/54Fe) between Fe(II)aq and Fe(III)-Si coprecipitates at circumneutral pH, as inferred by the three-isotope method, was ‒3.51 ± 0.20 (2σ) ‰ and ‒3.99 ± 0.17 (2σ) ‰ for coprecipitates that had Fe:Si molar ratios of 1:2 and 1:3, respectively. These results, when combined with earlier work, indicate that the equilibrium isotope fractionation factor between Fe(II)aq and Fe(III)-Si coprecipitates changed as a function of Fe:Si ratio of the solid. Isotopic fractionation was least negative when Fe:Si = 1:1 and most negative when Fe:Si = 1:3. This change corresponds with changes in the local structure of iron, as revealed by prior spectroscopic studies.
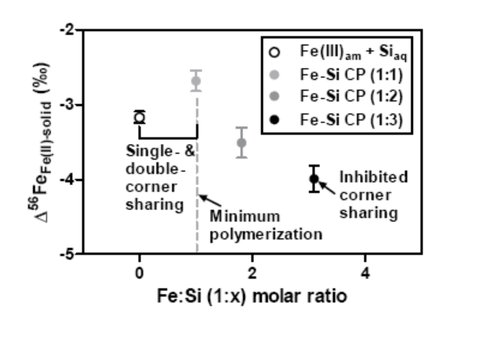
Experimentally determined equilibrium isotope fractionation factor (56Fe/54Fe) between Fe(II) and pure Fe(III)am (in Si-containing solution) and Fe-Si CP (in Si-free solution) (Wu et al., 2011), and Fe-Si CP1 and Fe-Si CP2 in this study, versus the Fe:Si molar ratio of the solid.
The above findings provide constraints on the range of equilibrium iron isotope fractionations that may be produced by different geochemical processes. In addition, when interpreted in a Fe mass-balance context, our findings provide strong support for DIR as a mechanism for producing Fe isotope variations observed in Neoarchean and Paleoproterozoic marine sedimentary rocks.
Figure 3 shows how low-δ56Fe values for Fe(II)aq can be produced by either extensive oxidation and precipitation of Fe(II)aq or partial reduction of Fe(III)am. The former produces
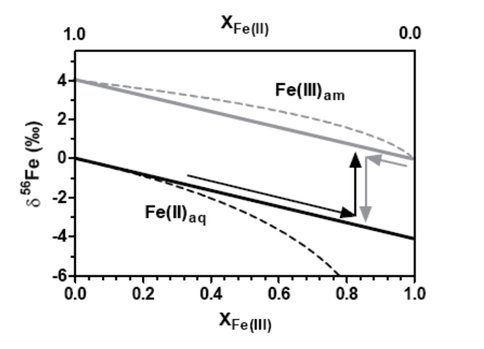
Schematic illustration of how low delta56Fe values for Fe(II)aq can produced by either extensive oxidation and precipitation of Fe(II)aq (XFe(II) going from 1 to near zero) or partial reduction of Fe(III)am (XFe(III) going from zero to a value lower than one).
small quantities of isotopically light Fe(II)aq, whereas the latter can generate a large quantity of isotopically fractionated Fe(II) upon separation of low-δ56 Fe(II) from residual Fe(III) in space or time. Solid lines indicate maximum equilibrium fractionation factor between Fe(II)aq and Fe(III)am as obtained in this study and dashed lines indicate a Rayleigh fractionation that has the same fractionation factor. These findings provide constraints for equilibrium fractionation factors between Fe(II)aq and Fe(III)am produced by different geochemical processes, which can in turn serve as a framework for interpreting iron isotope data in natural environments that contain reduced and oxidized iron.
Publications
-
Percak-Dennett, E. M., Beard, B. L., Xu, H., Konishi, H., Johnson, C. M., & Roden, E. E. (2011). Iron isotope fractionation during microbial dissimilatory iron oxide reduction in simulated Archaean seawater. Geobiology, 9(3), 205–220. doi:10.1111/j.1472-4669.2011.00277.x
-
Tangalos, G. E., Beard, B. L., Johnson, C. M., Alpers, C. N., Shelobolina, E. S., Xu, H., … Roden, E. E. (2010). Microbial production of isotopically light iron(II) in a modern chemically precipitated sediment and implications for isotopic variations in ancient rocks. Geobiology, 8(3), 197–208. doi:10.1111/j.1472-4669.2010.00237.x
-
Wu, L., Beard, B. L., Roden, E. E., & Johnson, C. M. (2009). Influence of pH and dissolved Si on Fe isotope fractionation during dissimilatory microbial reduction of hematite. Geochimica et Cosmochimica Acta, 73(19), 5584–5599. doi:10.1016/j.gca.2009.06.026
-
Wu, L., Beard, B. L., Roden, E. E., & Johnson, C. M. (2011). Stable Iron Isotope Fractionation Between Aqueous Fe(II) and Hydrous Ferric Oxide. Environ. Sci. Technol., 45(5), 1847–1852. doi:10.1021/es103171x
-
Wu, L., Beard, B. L., Roden, E. E., Kennedy, C. B., & Johnson, C. M. (2010). Stable Fe isotope fractionations produced by aqueous Fe(II)-hematite surface interactions. Geochimica et Cosmochimica Acta, 74(15), 4249–4265. doi:10.1016/j.gca.2010.04.060
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Brian Beard
Co-Investigator
Clark Johnson
Co-Investigator
Lingling Wu
Postdoc
Elizabeth Percak-Dennett
Doctoral Student
-
RELATED OBJECTIVES:
Objective 4.1
Earth's early biosphere.
Objective 7.1
Biosignatures to be sought in Solar System materials
