2010 Annual Science Report
 Massachusetts Institute of Technology
Reporting | SEP 2009 – AUG 2010
Massachusetts Institute of Technology
Reporting | SEP 2009 – AUG 2010
Genomic Relationships Among Basal Metazoans
Project Summary
Understanding the origins of animals (“Metazoa”) and the advent of metazoan complexity requires a proper understanding of the interrelationships among the living forms. To properly place animals like sponges and jellyfish into the tree of life, we have taken a multi-faceted approach using two different kinds of molecular data: traditional sequence-based molecular phylogenetics, and a new type of binary data, the presence or absence of specific microRNAs (short ~22 nucleotide non-coding RNA genes). Both data sets suggest that sponges are paraphyletic: some sponges are more closely related to jellyfish and humans than they are to other sponges (e.g., bath sponges). These results suggest that the last common ancestor of all living animals was organized like a true sponge, and thus our origins as complex animals lies within sponge biology.
Project Progress
The relationships at the base of the metazoan tree have been difficult to robustly resolve, and there are several different hypotheses regarding the interrelationships amongst sponges, cnidarians, ctenophores, placozoans and bilaterians, with each hypothesis having different implications for the body plan of the last common ancestor of animals and the paleoecology of the late precambrian. We have sequenced seven nuclear housekeeping genes from 17 new sponges, bringing the total to 29 species analyzed, including multiple representatives of the Demospongiae, Calcarea, Hexactinellida and Homoscleromorpha, and analyzed a data set also including six non-metazoan outgroups and 36 eumetazoans using a variety of phylogenetic methods and evolutionary models. We used leaf stability to identify rogue taxa and to investigate their effect on the support of the nodes in our trees, and we identified clades most likely to represent phylogenetic artifacts through the comparison of trees derived using different methods (and models) and through site-stripping analyses. Further, we investigated compositional heterogeneity and tested whether amino acid composition bias affected our results. Finally, we used Bayes factors to compare our results against previously published phylogenies. All our maximum likelihood and Bayesian analyses find sponges to be paraphyletic, with all analyses finding three extant paraphyletic sponge lineages, Demospongiae plus Hexactinellida, Calcarea and Homoscleromorpha. All but one of our Maximum likelihood and Bayesian analyses support the monophyly of Eumetazoa (here Cnidaria + Bilateria) and a sister group relationship between Placozoa (here Trichoplax adhaerens) and Eumetazoa. Bayes factors invariably provide decisive support in favor of poriferan paraphyly when compared against either a sister group relationship between Porifera and Cnidaria, or to a monophyletic Porifera with respect to a monophyletic Eumetazoa. Although we were able to recover sponge monophyly using our data set, this was only possible under unrealistic evolutionary models, if poorly performing phylogenetic methods were used, or in situations where the potential for the generation of tree reconstruction artifacts was artificially exacerbated. Everything considered, our data set does not provide any support for a monophyletic Diploblastica (here Placozoa + Cnidaria + Porifera) and suggests that a monophyletic Porifera may be better seen as a phylogenetic artifact.
To further test this result, we have pursued an independent avenue, namely the structure of microRNA pre- structures. Briefly, microRNAs (miRNAs) are small, ~22 nucleotide non-coding genes that negatively regulate protein-coding genes by binding with imperfect complementarity to sites in their 3’ untranslated regions (UTRs), thereby subjecting the transcript to cleavage or to blockage of its translation. Eumetazoan (the collective names for cnidarians + bilaterians) miRNAs are generally 70 nucleotide long structures, whereas plant and other eukaryotic miRNAs are much longer, usually between 120-200 nucleotides long. Interestingly, demosponge miRNAs are similar to plant miRNAs in being much longer than eumetazoan miRNAs. Because demosponge miRNAs appear to represent the primitive structure, we asked whether homoscleromorph and/or calcisponge miRNAs resembled eumetazoan miRNAs (and thus indicative of sponge paraphyly) or demosponge and plant miRNAs, consistent with sponge monophyly. To address this question small miRNA libraries were made from two calcisponges and one homoscler0moph and blasted against genomic sequences for two sponges, the calcisponge Sycon and the homoscleromorph Oscarella. The miRNA structures from Oscarella (but not Sycon) all resemble eumetazoan miRNAs, and not demosponge/plant miRNAs, consistent with the phylogenetic results supporting a close relationship between homoscleromorphs and eumetazoans to the exclusion of demosponges and calcisponges.
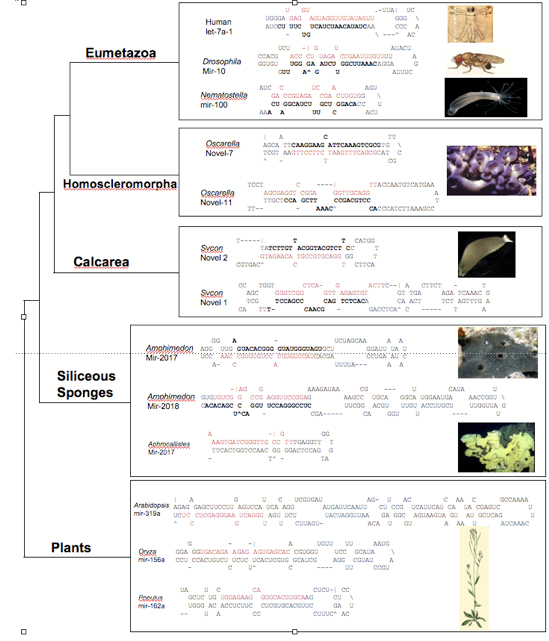
Representative pre-microRNA structures showing that the position of the mature miRNA sequence is found in close proximity to the terminal loop in eumetazoans and the homoscleomorph, while calcisponge, demosponge, and plant mature sequences vary widely in their position relative to the loop. Mature sequences are highlighted in red, star sequences are in bold. Pre-miRNA sequences for Eumetazoan, Demosponge, and Plant were obtained from miRBase. Sycon and Oscarella miRNAs were identified in small-RNA libraries and BLASTed against genome sequence available from recently sequenced genomes to obtain pre-sequences. All structures were predicted with the mFold program using default settings.
Publications
-
Sperling, E. A., Peterson, K. J., & Pisani, D. (2009). Phylogenetic-Signal Dissection of Nuclear Housekeeping Genes Supports the Paraphyly of Sponges and the Monophyly of Eumetazoa. Molecular Biology and Evolution, 26(10), 2261–2274. doi:10.1093/molbev/msp148
-
Sperling, E. A., Robinson, J. M., Pisani, D., & Peterson, K. J. (2010). Where’s the glass? Biomarkers, molecular clocks, and microRNAs suggest a 200-Myr missing Precambrian fossil record of siliceous sponge spicules. Geobiology, 8(1), 24–36. doi:10.1111/j.1472-4669.2009.00225.x
-
PROJECT INVESTIGATORS:
-
PROJECT MEMBERS:
Maja Adamska
Co-Investigator
Davide Pisani
Co-Investigator
Scott Nichols
Collaborator
Jeffery Robinson
Doctoral Student
Erik Sperling
Doctoral Student
-
RELATED OBJECTIVES:
Objective 4.2
Production of complex life.
