2014 Annual Science Report
 University of Illinois at Urbana-Champaign
Reporting | SEP 2013 – DEC 2014
University of Illinois at Urbana-Champaign
Reporting | SEP 2013 – DEC 2014
Executive Summary
This is a progress report from the University of Illinois NAI Team on the Project “Towards Universal Biology: Constraints from Early and Continuing Evolutionary Dynamics of Life on Earth”, and covers the highlights of our research in the time period from January 1 2013 to December 2014. The organization of this report parallels the structure used in our Annual Report Year 1 and of the forthcoming official Annual Report Year 2.
The specific objectives of our research are the following four Themes which are, in brief: [1] Theoretical understanding of the universal features governing living systems, their operation, evolution and origin; [2] Constraints on the nature of life before the Last Universal Common Ancestor (LUCA), in particular to identify new signatures of the collective state of life (“progenote”) which enabled the evolution of the cell to occur so rapidly; [3] To explore the breakdown of the progenote and the transition to vertical evolution; [4] Explore the interplay between biological and environmental determinants of the rate of evolution.
We have made significant progress in each of these areas, organized along the lines of the following Projects.
Project 1: Dynamics of Self-Programming Systems. This project is a theoretical attempt to understand how evolution can arise from inanimate physical systems. The key idea is that matter can organize into structures that not only replicate and carry information, but are able to program and reprogram themselves functionally. We have already been able to construct simple computer programs that can increase their complexity in an open-ended way, but in this grant period we have been building a mathematical formulation of how this arises using recursive function theory. [Goldenfeld, DeVille]
Project 2: Cells as engines and the serpentinization hypothesis for the origin of life. We are attempting to understand the thermodynamics of living cells, in particular the way in which cells perform work. Although very complicated in detail, cells essentially function as a rather baroque heat engine, using proton gradients to drive the storage and usage of energy as ATP and ADP. In particular, they convert disequilibria in proton gradients to disequilibria between ATP and its hydrolysis products (ADP and Pi). This process is not only a universal feature of contemporary life, but, as Russell and Branscomb have argued, is a natural mechanism for life to exploit if it began in serpentinization environments, such as those in alkaline hydrothermal vents. We have recently formulated a description of this universal cellular engine using statistical mechanics, enabling us to understand in detail how proton disequilibria drive and constrain cellular function, and the efficiency of this process. Our model is the first statistical thermodynamic description of the fundamental energetics of living cells, taking into account the fundamental role of disequilibrium conversion. The models of this process contain adjustable parameters for rate processes, and the next step of our research will be to understand how these parameters have been chosen by nature. Our idea is that the optimal values of the parameters are selected by evolution, and so we plan to do computer simulations of the evolution of communities of thermodynamic engines, to see which are selected under which environmental conditions. Our work will help constrain the environments on Earth under which life probably evolved, in particular the hypothesis that geochemical serpentinization provided the natural setting under which the proton gradient driven cellular engine could have arisen and form the core of all living organisms on Earth. [Branscomb, Goldenfeld]
Project 3: The origin of homochirality. A universal aspect of living systems on Earth is their homochirality: Life uses dextrorotary sugars and levorotary amino acids. The reasons for this are hotly debated and not close to being settled. However, the leading idea is that autocatalytic reactions grew exponentially fast at the origin of life, and whatever chiral symmetry breaking was accidentally present became amplified subsequently. For this argument to be viable, it has to be shown that autocatalysis can indeed amplify homochirality. Secondly, one must consider the potentially confounding effects of spatial extension, which could give rise to spatial heterogeneity in the pattern of chiral symmetry breaking. We have succeeded in providing an improved formulation of the famous homochirality model of F.C. Frank, one that removes an unphysical requirement and moreover includes the inevitable and important stochasticity arising from chemical reactions. The mathematics shows that homochirality can arise by a previously unnoticed mechanism – the stabilization of homochirality into states that are least affected by noise. This mechanism is different from potential-based mechanisms for bistability, because it is the noise itself that generates the symmetry breaking. We have also succeeded in formulating this stochastic model with spatial extension. Our preliminary result is that the autocatalytic mechanism for homochirality does not seem to be stable in space: life would self-organize into local domains where homochirality was present, but of a different sign in neighbouring domains. If our ongoing research confirms this result, then it will cast doubt on the autocatalytic proposal for homochirality. [Goldenfeld, DeVille, Branscomb]
Project 4: Rapid evolution in stressed populations: theory. A key problem in astrobiology is what governs the rate of evolution, and how is this rate influenced by the environment? This question is being explored experimentally in Theme 4 from our original proposal. In tandem with the ongoing experiments on bacterial and archaeal stress response in microfluidic engineered environments, we have performed a detailed stochastic individual level calculation of the way in which environmental fluctuations can drive ecological processes and vice versa. The main result from this work is that evolutionary timescales can be accelerated significantly and indeed may occur on the same scale as environmental variability, not much longer as is conventionally assumed. The specific setting for this work was an ecosystem comprising predators and prey, but where it was possible for a mutant prey strain to arise, one which is both less able to compete with the wild-type for nutrient and is also less nutritious to the predator. We showed that number fluctuations in the community drive large and unusual population cycles. These cycles have strange phase relationships between the predator and prey species, and these are hallmarks of population dynamics which is strongly influenced by evolution. Our work showed that these phenomena are rather universal, and do not require rather detailed and specific forms of functional response, as has been proposed up to now. These findings provide a framework that can explain the rapid evolutionary dynamics that has been reported in laboratory experiments, and which are the subject of Theme 4. [Goldenfeld, Cann, Fouke, Mackie, Werth]
Project 5: The origins of life’s diversity. The huge diversity of life poses a major challenge to ecological theory and a major source of optimism for astrobiology. Ecological theory argues that a single environmental niche should be colonized by a single species of organism, or perhaps a small community, and so the diversity of life should be essentially a measure of the number of niches present. The dramatic failure of this idea has been formulated in what is sometimes known as “The Paradox of the Plankton” and is fundamentally unresolved at the present time. The huge diversity of life does suggest, however, that the ability of life to explore, colonize and especially create environmental niches has been drastically underestimated. Accordingly, the likelihood of extraterrestrial life arising is also underestimated, or at least inadequately estimated, by our present understanding of biological evolution. We have started to address this question theoretically, prompted initially by our ongoing work on the breakdown of the progenote state. We have succeeded in developing a theoretical approach for predicting biodiversity in multi-dimensional niche spaces, arising due to ecological drivers such as competitive exclusion. The novelty of our approach relies on the fact that ecological niches are described by sequences of strings, which allows us to describe multiple traits. Our work proposes a mathematical framework for analyzing pattern forming instabilities in these models, and shows surprisingly that the analytic linear theory predicts the asymptotically long time population distributions of niches in the model. We also proposed a test for identifying ecological drivers in biodiversity distributions, based on representing ecosystem data by means of a certain transform introduced in the theory. The theoretical work in this project models the formation of species or genetic clusters of organisms as occurring via a pattern forming instability in genotype or phenotype space. An initially uniform distribution of organisms or genomes spontaneously self-organizes into clusters that one can think of as species or operational taxonomic units. We believe that the mathematical techniques developed here also underpin the breakdown of a progenote state, stabilized by massive horizontal gene transfer, into a state where genetic clusters form corresponding to the domains of life. Our work shows that this can happen, in principle, and in the coming grant period will be used to attack the question of how the domains of life arise from the most unstable modes of the progenote state. [Goldenfeld, DeVille]
Project 6. Mining Archaeal Genomes for Signatures of Early Life. In this project we are attempting to analyze the evolution of the central cellular functions of translation, transcription, replication, and metabolism. In the case of metabolism we are initially focusing on those reactions leading to different bioenergetics pathways. Methanogenic archaea have the unique ability to generate methane from simple carbon substrates like acetate and methylamines in anaerobic environments typical to early life on earth and perhaps other planets. To process each substrate, the organisms had to evolve unique pathways with vastly different thermodynamic behavior. To date we have characterized the physical properties and constructed a kinetic model for the methanogenesis pathways in the methanogenic archaeum Methanosarcina acetivorans. M. acetivorans can process a large number of substrates that are taken up individually in other diverse, but related methanogens. In this model, 26 reactions in the methanogenesis pathways are coupled to a cell mass production reaction that updates enzyme concentrations. RNA expression data (RNA-seq) measured for cell cultures grown on acetate and methanol are used to estimate relative protein production per mole of ATP consumed. A draft transcriptional regulation network based on the RNA-seq and mRNA life time studies is nearly completed which will allow dynamic regulation. [Luthey-Schulten, Cann]
Project 7. Control of evolvability and chromosomal rearrangement by stress. We have screened for suppressors of the requirement for the RpoS stress-response in chromosomal rearrangement by using a transposon with outward-facing promoters that activates genes in the absence of stress. This could tell which stress-induced function is required for chromosomal rearrangement. Most of the suppressors isolated have activated parts of cryptic prophages: bacterial viruses that have lost their ability to lyse the host cell, and so the genetic material lives on in an inactive form in the host cell’s DNA. Four of these suppressors have resulted in the activation of alternative recombination through the RecET pathway. We are testing whether these have bypassed the requirement for stress, rather than suppressing it. The most interesting isolate at present has the transposon disrupting the gene dgt, which encodes a deoxyguanidine triphosphohydrolase. This protein regulates both deoxyguanidine triphosphate and thymidine triphosphate pools, and is stress-induced. Other work has shown that DNA nucleotide pool content modulates mutation rates, but the mechanism for this is unknown. Work on validating and explaining the effect of dgt is ongoing.
Nucleoid-associated proteins (NAPs) both modify the structure of bacterial chromatin and act as global regulators of gene expression. We have completed a study of the involvement of nucleoid-associated proteins in stress-induced mutation, finding that the effects of four NAPs are explained by factors other than their ability to modify chromatin structure. Two of the four modify the availability of oxygen radicals, another regulates the expression of the DNA damage response, specifically the availability of trans-lesion polymerases, and the fourth is required for maintenance of the conjugative plasmid that carries the assay system.
We have established the unexpected result that both spontaneous mutation and chromosomal rearrangement, both stress-induced and growth-dependent, require 8-oxoguanine incorporation into DNA. We already knew that double-strand breaks are required, but the requirement for oxidative DNA lesions is not via double-stand break formation. Using genetic control of responses to DNA damage via oxidation and using chemical scavengers of reactive oxygen, we show beyond doubt that there is a requirement for persistent 8-oxoguanine incorporation. When we over-express enzymes that specifically excise 8-oxoguanine from DNA, point mutation formation is deficient. Hence the lesion must remain in the DNA to be effective, and so mutation is presumably formed by induction of trans-lesion synthesis. We have not yet shown that chromosomal rearrangement requires the lesion to remain in DNA, but are setting up a new assay to test that. Because reactive oxygen will not be present in all environments, we tested whether other DNA lesions could substitute for 8-oxoguanine. Suppressing reactive oxygen with thiourea, a condition that allows no mutation, we have shown that we can restore mutation with alkylating agents or with ultraviolet irradiation. Genetic tests indicate that these treatments restored the same stress-dependent pathway that we study. We are now investigating whether the lesion-induced stress-dependent mutation occurs because stress allows more uptake of altered bases into DNA, or whether the lesions allow increased use of error-prone DNA polymerases. [Hastings, Rosenberg]
Project 8: Culturing microbial communities in controlled stress micro-environments. This project advances the objectives of Theme 4B: to explore adaptation and evolution of organisms under constraint environmental conditions, and to compare the behavior across two Domains of Life in order to identify and quantify universal aspects of evolutionary response, if present. To accomplish this, our team has designed and built the GeoBioCell —- a microfluidic gradient chamber created by etching a 1200-well array and boundary channels into a silicon wafer using photolithography methods. Archaea Methanosarcina acetivorans and Bacteria Escherichia coli were chosen as the model organisms to evaluate, and constraint conditions were created by establishing a concentration gradient within the GeoBioCell of an inhibitory antibiotic, i.e., puromycin for M. acetivorans and ciprofloxacin for E. coli. Cell growth was evaluated in batch systems for both M. acetivorans and E. coli in the presence of the antibiotic to determine the minimum inhibitory concentrations (MICs). The GeoBioCell design and results of E. coli growth in the presence of a ciprofloxacin are illustrated in Figure 1. Substrates (LB media, dissolved O2) were continuously delivered to the boundary channels of the GeoBioCell, while ciprofloxacin was continuously delivered to only the bottom boundary channel to establish a concentration gradient. E. coli cells were inoculated into the center port; they are labeled with a green fluorescence protein (GFP) so growth was tracked using fluorescent microscopy. Cells began to visibly grow and spread within hours, and after 30 hours (Figure 1) E. coli have explored the entire 1200-well array and are primarily growing in wells along the upper boundary channel where ciprofloxacin concentration is lowest. There is also some E. coli in wells along the bottom left channel, due to dilution of ciprofloxacin as it flows from right to left through the boundary channel. Under higher magnification (Figure 1), individual cells are observable in a single well. We note that along the bottom boundary toward the inlet individual cells were also visible, but they were not fluorescing (implying they are not replicating). No adaptation to ciprofloxacin was observed in this experiment, even after two weeks. We hypothesis that this is due to limited O2 diffusion from the boundary channels, and have redesigned the GeoBioCell with a gas permeable coverslip to allow O2 diffusion directly into all 1200-wells in our next experiment. [Cann, Fouke, Mackie, Werth]
Project 9A: Evolution through the lens of codon usage: frequent horizontal gene transfer extending beyond species. Over 30 years ago, the codon usages of genes in a genome were interpreted in terms of (i) typical codon usage, (ii) codon usage of abundant proteins (adapted to higher expression levels), and (iii) other maladapted codon usages. In 1991, Médigue et al. recognized that many of these maladapted Escherichia coli genes define a third distinct codon usage type, and noted that many of these genes had been acquired by horizontal gene transfer (i.e., they are alien genes). It has been assumed that the distinctive codon usages reflect those of phylogenetically distant donors, and that with time the alien codon usage of an acquired gene would drift (ameliorate) to match those of the host organism. Using comparative genome analysis, we demonstrated that (i) the codon usage of the most recently acquired genes are indeed distinct, (ii) their codon usage is found only in the genomes of closely related species (i.e., it is not that of a hypothetical distantly related species), (iii) the recently acquired gene codon usages are so similar between E. coli and Salmonella enterica that these species must be drawing upon the same pool of donor genomes, and (iv) that, taken together, these and other closely related species must constitute the reservoir from which the plurality of their recently acquired genes are drawn. This is largely consistent with recent perspectives on the pangenome of species, but it requires that frequent sharing extend substantially beyond current species circumscriptions, which led us to propose a supraspecies pangenome). Neither the pangenome hypothesis, nor our extension of it, provides an explanation for the distinctive codon usage of the recently acquired genes; if they are moving amongst closely related genomes they would be expected to drift to match some average codon usage of those genomes, but they do not. [Dawson, Olsen, Whitaker]

Project 9B: Evolution through the lens of codon usage: vertically inherited genes resist amelioration. The codon usage originally recognized due to its occurrence in recently acquired genes can also be found in genes that have been present in genomes for many 10’s of millions of years. For example, the major “Salmonella Pathogenicity Islands” (SPIs) of S. enterica have been stably present in the genomes since the origins of this species, and some even longer. It had been previously noted that in spite of this, they do not match the G+C content or codon usages of typical or highly expressed genes of the genome. If these genes have been vertically inherited throughout the history S. enterica, they would be expected to drift to match the properties of the rest of the genome, but they have not. Further, we have demonstrated that their codon usages are the same as those of the most recently acquired genes, suggesting a common basis for their distinctive properties. [Dawson, Olsen, Whitaker]
Project 9C: Evolution through the lens of codon usage: alien gene codon usage is an intrinsic adaptation to stress. It appears that the recently transferred genes and the SPIs are subject to some selective force that acts upon their codon usage. We have shown that is not just a consequence of their G+C content, and it is not just moving the opposite direction from high-expression codon usage. It appears that these genes are subject to positive selection for a third category of codon usage. In seeking clues as to the nature of the selection, the most striking data sets relate to the ppGpp and the stringent response. In textbook molecular biology, the stringent response is responsible for a major shutdown of cell biosynthetic machineries in times of starvation. Two published datasets provide gene expression data under stringent response conditions: one in early stationary phase and one in late stationary phase. Although focus is usually on the things that the stringent response shuts down, the genes most up-regulated (≥10-fold) by this starvation induced response also have the codon use of the SPIs and the recently transferred genes. These data are not fully independent in that the stringent response does induce some of the SPIs. However, stress, and possibly more specifically starvation, are recurring themes when examining the biology of the genes of these codon usages. A starvation codon usage has been previously proposed in the literature, though generally from a more theoretical perspective, and some aspects of our data differ from their predictions. [Dawson, Olsen, Whitaker]
Project 9D: Evolution through the lens of codon usage: starvation codon usage is recurring in nature. We have not completed comprehensive analyses, but many to most of the available genome sequences for Bacteria and Archaea show evidence of a third distinct codon usage (in addition to typical and high-expression). Preliminary analyses of the archaeon Sulfolobus islandicus have led to an additional collaborative project between Whitaker and Olsen in which the codon usage of genes involved in cellular responses to stress induced by viral infection are being investigated. We have not yet examined the genomes of Eucarya. [Dawson, Olsen, Whitaker]
Project 9 Tools: All of the tools that we have developed for this work are being integrated into the RAST genome annotation system at Argonne National Laboratory. [Olsen]
Project 10: Development of molecular genetic tools in the crenarchaeote Sulfolobus islandicus. One of the primary steps toward understanding evolutionary steps at the Darwinian threshold of cellular individuality involves identifying the molecular components of early cells. Our goal is to take a comparative genomic approach to define the molecular machinery that differentiate the Bacterial from its sister lineage that later diverged to became the Archaea and Eukaryotes. One of the obstacles clouding our view of these early cells from a comparative approach is the large number of conserved hypothetical genes present in Archaeal and Eukaryote genomes whose cellular functions are unknown. Over the past year we have developed a random transposon insertion library with the goal of identifying conserved hypothetical genes, whose cellular functions are essential. To begin with we developed a genetic marker with strong positive selection for agmatine prototrophy. Using selection for this marker we constructed a transposon mutagenesis system and tested for random insertions by selecting and sequencing insertions. In order to saturate the genome and identify essential genes we estimate that we need to select 20,000 individual random insertions in the 2.59 Mbp genomic background of M.16.4. Great progress has been made toward this goal. We currently have nearly 7,000 clones isolated and estimate that our complete ordered random library will be constructed and sequenced by February 2015.
We anticipate that two initial publications will result from this work, one describing the essential gene in this model crenarchaeon and one describing the distribution of insertions sites as it relates to genome architectures. Future work using this library will screen for novel conserved hypothetical genes associated with replication, recombination, and repair and other central DNA processing functions in the archaeal cell. [Dawson, Whitaker, Olsen]
Project 11: Isolation and genomic sequencing of novel eukaryotic amoebae from freshwater and marine environments. The immense diversity of microbial eukaryotes is not reflected in current or completed genome projects. Sequencing efforts have focused overwhelmingly (over 86%) on either opisthokont (animal and fungi) and plant lineages, or secondarily-reduced parasitic protists. Few genomes of free-living microbial eukaryotes are sequenced, despite their critical importance in ecology, evolution, and basic cellular biology. By comparing the genomes of these new protists with known organisms, we will gain insight into the diversity and complexity of the eukaryotic cell. We can also infer key evolutionary events in the history of the eukaryotes.
As with most bacteria, most free-living protistan taxa are difficult to obtain in pure culture, and are deposited in culture collections as mixed consortia with bacteria. To aid in study of early eukaryotic evolution, we are have cultured about 120 novel amoeba isolates from from marine and freshwater environments. Many of the newly classified isolates (6/33) represent novel groups of eukaryotes. These amoebae range in size from about 3 µM to over 100 µM and have diverse types of motility. Thirty-three have been sequenced and classified using ssu rRNA. None of the isolates have close relatives with sequenced genomes. [Dawson, Whitaker, Olsen]
Project 12: Enrichment of eukaryotic DNA in mixed cultures of eukaryotic genomic DNA sequencing. The main limitation in sequencing genomes from diverse eukaryotic microbes is that they are heterotrophic and are grown with bacteria. We are developing several methods to purify eukaryotic DNA from bacterial DNA deriving from organisms with which they are co-cultured. First, because many amoebae from cysts, we are able to encyst the protists, and harvest and wash bacteria from the cyst prepration. This process has worked well with Nuclearia, and we are currently having this genome sequenced at U Illinois. The second method involves using affinity purification using chromatin-immunoprecipitation to enrich eukaryotic from bacteria DNA in mixed samples. We have preliminary support for that this method enriches for eukaryotic DNA for a novel isolate of marine amoeba (P5A) that does not encyst. This method would allow us to access any eukaryote in culture for genomic sequencing. [Dawson, Olsen, Whitaker]
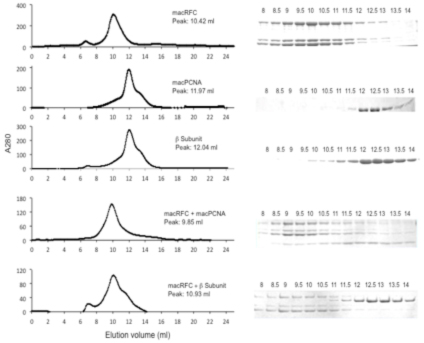
Project 13: Experimental determination of the existence of the Darwinian Transition. This project aims at determining whether translation, transcription, and replication have crossed the Darwinian threshold, defined as a stage at which the molecular machinery involve in each of the three processes fails to accept a component from a different domain. Our initial experiment targets DNA replication by examining interchangeability of the DNA replication processivity factor known as the sliding clamp. It is only by interacting with the sliding clamp that DNA polymerases in extant organisms can gain the speed required to replicate their large genomes. We have expressed and purified a sliding clamp from each of the three domains of life E. coli d-subunit, M. acetivorans PCNA, and human PCNA). Each sliding clamp is a circular polypeptide and therefore requires a specialized protein known as the clamp loader to break it open and load it onto DNA to facilitate interactions with the replicative DNA polymerase. There are three stages in clamp loading: interaction of the clamp loader with the sliding clamp, opening of the sliding clamp by the clamp loader and finally sealing of the sliding clamp onto DNA. We have tested the capacity of an archaeal clamp loader (M. acetivorans RFC) to interact with its cognate sliding clamp (MacPCNA) and a sliding clamp from a different domain of life, i.e., E. coli b-subunit. The method applied used analytical gel filtration chromatography, where individual proteins and mixture of proteins were injected into a Sephadex column to monitor interaction between the clamp loader and the sliding clamps. The results are presented in figure 1 as chromatograms and protein gels (SDS-PAGE). The peak elution volume of MacRFC was ~10.42 ml (215 kDa), while the sliding clamps eluted at ~12 ml for both b-subunit and MacPCNA (80 kDa). A mixture of MacPCNA and MacRFC on the other hand eluted at a peak of 9.85 ml, showing that MacRFC interacts with its cognate sliding clamp (MacPCNA). Mixing of the bacterial sliding clamp, i.e., E. coli b-subunit with the archaeal clamp loader (MacRFC) did not change the elution volume of the two proteins. All gel filtration results were confirmed with protein gels. Thus based on these experiments, the archaeal clamp loader does not recognize the bacterial sliding clamp. Since the sliding clamp recruits the DNA replication protein complex and is therefore central to DNA replication, our results suggest that replication in bacteria has crossed the Darwinian threshold. We are currently using a different method based on primer extension or DNA synthesis to confirm or disprove our findings.
Education and Public Outreach. The E/PO for the Illinois-Baylor-UC Davis NAI has remained extremely active and has successfully fulfilled all of the mission goals for Year 2. These activities have included:
(1) K-12 Formal Education: Professor Lizanne DeStefano, Director of the Illinois iSTEM initiative (http://www.istem.illinois.edu), has joined our NAI E/PO. We have established a Student Teacher Scientist Partnership (STSP) with Urbana Middle School (UMS) and Urbana High School (UHS), and will expand to include other public school districts in 2015-16. In February 2015 our STSP program was formally approved by the Urbana School District (USD) Superintendent and the principles of both UMS and UHS. Middle school and high school science teachers were then recruited via information sessions at UMS and UHS. From an original group of 40 teachers, 12 were chosen to directly participate in two NAI Universal Biology Teacher Workshops that were held at the Institute for Genomic Biology (IGB) on the Illinois campus and lead by Fouke and DeStefano. The workshop goals are to produce, evaluate, refine, and disseminate hands-on field based instructional units built around the Astrobiology Roadmap and will be directly aligned with Next Generation Science Standards (NGSS) content. These new curricula will be implemented in the UMS and UHS classrooms in Spring 2015 and Fall 2015. Three more Universal Biology Teacher Workshops are planned for Spring 2015 at the IGB, which will include field trips to the Field Museum and Adler Planetarium in Chicago. In addition, teachers are required to complete the NAI MOOC (described below) and receive formal continuing education credits for the Urbana School District.
(2) MOOC Web-Based Education: A new Massively Open On-Line Course (MOOC) entitled Emergence of Life was created during Year 1 and was offered twice during Year 2 (see http://go.illinois.edu/emergenceoflife). In Summer 2014 the MOOC had 23,000 students enroll that represented 154 countries around the world. The MOOC was again offered in Fall 2015 and an additional 13,000 students enrolled from 101 different countries. We plan to regularly offer the MOOC each year and each MOOC offering is being quantitatively assessed. The MOOCs involved members of the NAI team at all levels ranging from students to professors.
(3) Higher Education: The NAI MOOC has been modified and turned into a formal “for credit” Illinois College of Liberal Arts and Sciences (LAS) On-Line course entitled GEOL 111 Emergence of Life. The course was first offered in Spring 2014 and had 82 students enrolled, and will be offered again in the Spring 2015 semester.
Fouke has offered in both Year 1 and Year 2 a new Illinois Campus Honors Program (CHP) course entitled CHP 395 Yellowstone Astrobiology. The course includes classroom lectures and laboratory experiences throughout the Fall semester, and included which a capstone field course in late November in which 20 Illinois CHP students are brought to Yellowstone to complete and report on astrobiology-driven field experiments. Werth co-taught the course in Year 2 and helped teach the Yellowstone NAI field components.
(4) Informal Education – Fouke is offering a combined field and classroom course for the general public on January 7-9, 2015, in Yellowstone National Park. The course is offered through the Yellowstone Association Institute (YAI; https://www.yellowstoneassociation.org) and is entitled Yellowstone Astrobiology: Universal Biological Connections. Enrollment is capped at 25 and is composed of professionals of all ages and from all walks of life across the United States. The field component will be taught on snowshoes and cross-country skis.
(5) Book – Fouke is writing a new Yellowstone Astrobiology textbook to be used with the NAI MOOC and all associated formal and informal on-line and in-person Illinois NAI courses. This book will be published by the University of Illinois Press and is a collaborative effort between Fouke and Tom Murphy, acclaimed Yellowstone photographer (http://www.tmurphywild.com). The target date for completion of the book is Summer 2015.

Publications
-
Boyd, V., Yoon, H., Zhang, C., Oostrom, M., Hess, N., Fouke, B., … Werth, C. J. (2014). Influence of Mg2+ on CaCO3 precipitation during subsurface reactive transport in a homogeneous silicon-etched pore network. Geochimica et Cosmochimica Acta, 135, 321–335. doi:10.1016/j.gca.2014.03.018
-
Peterson, J. R., Labhsetwar, P., Ellermeier, J. R., Kohler, P. R. A., Jain, A., Ha, T., … Luthey-Schulten, Z. (2014). Towards a Computational Model of a Methane Producing Archaeum. Archaea, 2014, 1–18. doi:10.1155/2014/898453
-
Russell, M. J., Barge, L. M., Bhartia, R., Bocanegra, D., Bracher, P. J., Branscomb, E., … Kanik, I. (2014). The Drive to Life on Wet and Icy Worlds. Astrobiology, 14(4), 308–343. doi:10.1089/ast.2013.1110
-
Shih, H-Y., & Goldenfeld, N. (2014). Path-integral calculation for the emergence of rapid evolution from demographic stochasticity. Phys. Rev. E, 90(5), None. doi:10.1103/physreve.90.050702